

Bioprocess Engineering, Transcriptome, and Intermediate Metabolite Analysis of L-Serine High-Yielding Escherichia coli W3110


Abstract
:1. Background
2. Materials and Methods
2.1. Strains, Media, and Materials
2.2. Construction of Gene Knockout Mutants
2.3. Construction of the glyA Mutant Library
2.4. Chromosomal Integration of Glymut Constructs
2.5. Plasmid Construction for Overexpression of L-Serine Biosynthetic Pathway Components
2.6. PGDH and SHMT Enzyme Assays
2.7. Shake Flask and Fed-Batch Fermentation
2.8. Sample Preparation and Extraction for Intermediate Metabolite Analysis
2.9. Analytical Methods
2.10. Transcriptome Datasets
3. Results and Discussion
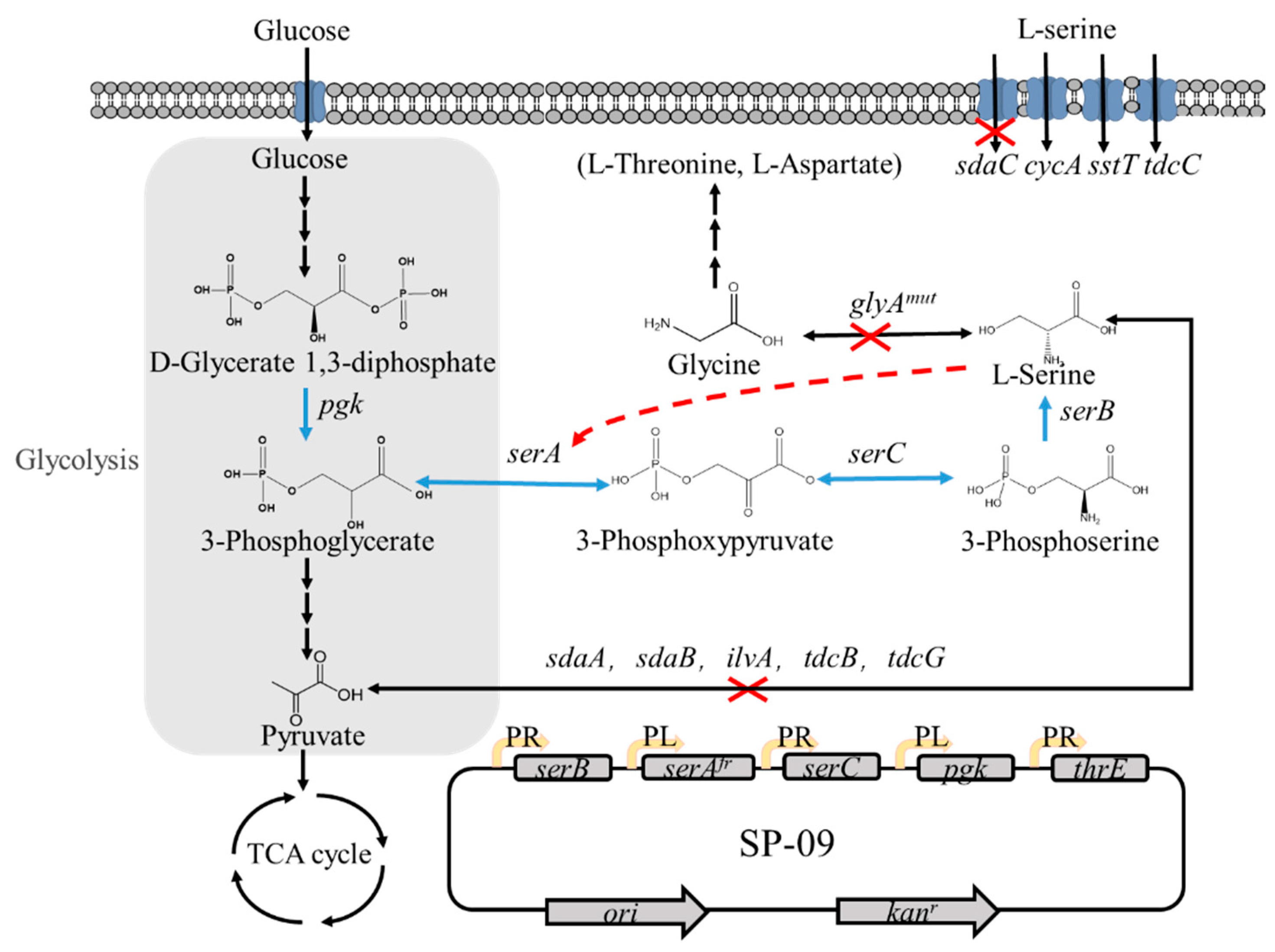
3.1. Construction of the L-Serine Production Strain from E. coli W3110
3.2. Influence of Mutations in glyA on L-Serine Production and Cell Growth
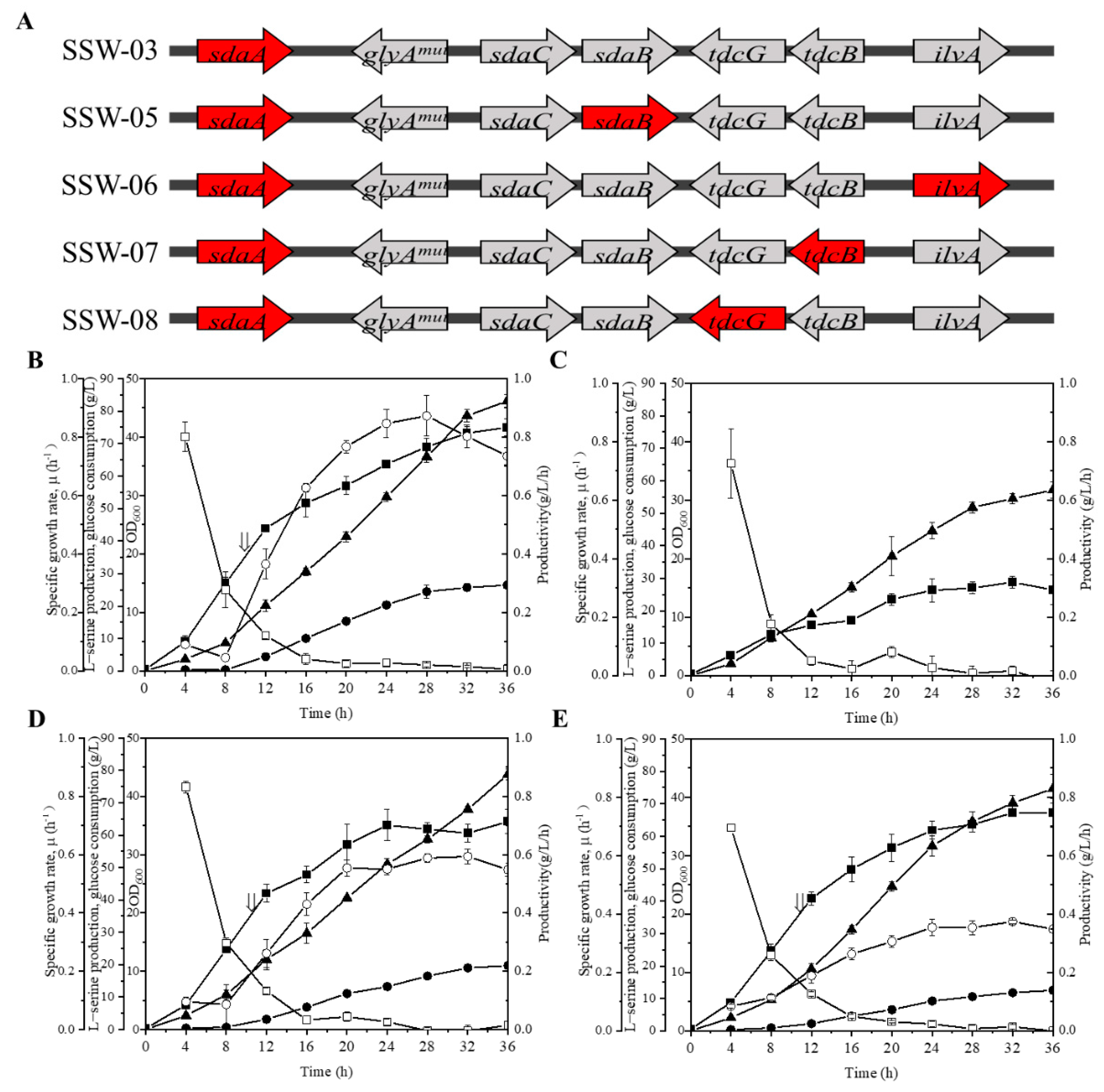
3.3. Influence of sdaB, ilvA, tdcB and tdcG Deletion on L-Serine Production
3.4. Effect of Engineering L-Serine Transport System on Strain Productivity
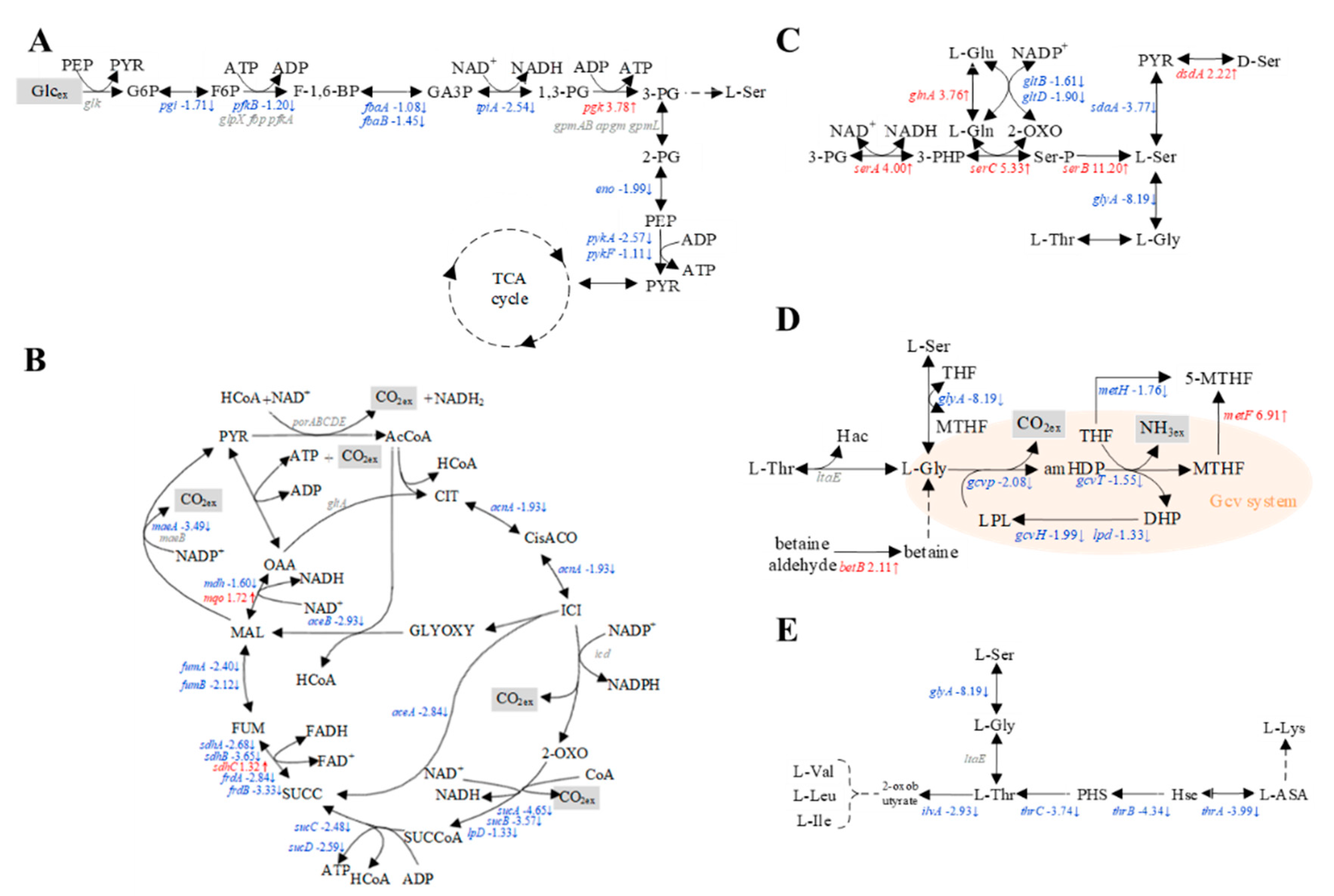
3.5. Transcriptomic Analysis of E. coli W3110 and SSW-10/SP-09
3.6. Intermediate Metabolite Analysis of E. coli W3110 and SSW-10/SP-09
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Glcex | Extracellular glucose |
| PEP | Phosphoenolpyruvate |
| PYR | Pyruvic acid |
| G6P | Glucose-6-phosphate |
| F6P | Fructose-6-phosphate |
| F-1 | Fructose-1 |
| 6-BP | 6-bisphosphate |
| GA3P | Glyceraldehyde-3-phosphate |
| 3-BG | 3-bisphosphoglycerate |
| 3-PG | 3-phosphoglycerate |
| 2-PG | 2-phosphoglycerate |
| AcCoA | Acetyl-coa |
| Cit | Citric acid |
| cis-Aco | Cis-aconitic acid |
| ICit | Isocitric acid |
| 2-OXO | 2-oxoglutarate |
| SucCoA | Succinyl-coa |
| Suc | Succinic acid |
| Fum | Fumaric acid |
| Mal | Malic acid |
| OAA | Oxaloacetic acid |
| NAD+ | Oxidized nicotinamide adenine dinucleotide |
| 3-PHP | 3-phosphonooxypyruvate |
| L-Ser | L-serine |
| D-Ser | D-serine |
| L-Leu | L-leucine |
| L-Ile | L-isoleucine |
| L-Val | L-valine |
| Gly | Glycine |
| L-Gln | L-glutamine |
| L-Glu | L-glutamate |
| PLP | Pyridoxal 5-phosphate monohydrate |
| THF | Tetrahydrofolate |
| MTHF | 5,10-methylene tetrahydrofolate |
| LPL | Lipoylprotein |
| DHP | Dihydrolipoylprotein |
| amDHP | Aminomethyldihydrolipoylprotein |
References
- Mundhada, H.; Schneider, K.; Christensen, H.B.; Nielsen, A.T. Engineering of High Yield Production of L-Serine in Escherichia coli. Biotechnol. Bioeng. 2016, 113, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Leuchtenberger, W.; Huthmacher, K.; Drauz, K. Biotechnological production of amino acids and derivatives: Current status and prospects. Appl. Microbiol. Biotechnol. 2005, 69, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Werpy, T.A.; Holladay, J.; White, J.F. Results of Screening for Potential Candidates from Sugars and Synthesis Gas. In Top Value Added Chemicals From Biomass; Pacific Northwest National Laboratory: Richland, WA, USA, 2004; p. 76. [Google Scholar]
- Wendisch, V.F. Metabolic engineering advances and prospects for amino acid production. Metab. Eng. 2020, 58, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xu, G.; Shi, J.; Koffas, M.A.G.; Xu, Z. Microbial Production of l-Serine from Renewable Feedstocks. Trends Biotechnol. 2018, 36, 700–712. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.; Wittmann, C. Systems and synthetic metabolic engineering for amino acid production—The heartbeat of industrial strain development. Curr. Opin. Biotechnol. 2012, 23, 718–726. [Google Scholar] [CrossRef]
- Chao, H.; Wu, B.; Shen, P. Overexpression of the methanol dehydrogenase gene mxaF in Methylobacterium sp. MB200 enhances L-serine production. Lett. Appl. Microbiol. 2015, 61, 390–396. [Google Scholar] [CrossRef]
- Peters-Wendisch, P.; Netzer, R.; Eggeling, L.; Sahm, H. 3-Phosphoglycerate dehydrogenase from Corynebacterium glutamicum: The C-terminal domain is not essential for activity but is required for inhibition by L-serine. Appl. Microbiol. Biotechnol. 2002, 60, 437–441. [Google Scholar] [CrossRef]
- Peters-Wendisch, P.; Stolz, M.; Etterich, H.; Kennerknecht, N.; Sahm, H.; Eggeling, L. Metabolic engineering of Corynebacterium glutamicum for L-serine production. Appl. Environ. Microbiol. 2005, 71, 7139–7144. [Google Scholar] [CrossRef]
- Stolz, M.; Peters-Wendisch, P.; Etterich, H.; Gerharz, T.; Faurie, R.; Sahm, H.; Fersterra, H.; Eggeling, L. Reduced folate supply as a key to enhanced L-serine production by Corynebacterium glutamicum. Appl. Environ. Microbiol. 2007, 73, 750–755. [Google Scholar] [CrossRef]
- Zhu, Q.; Zhang, X.; Luo, Y.; Guo, W.; Xu, G.; Shi, J.; Xu, Z. L-Serine overproduction with minimization of by-product synthesis by engineered Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 2015, 99, 1665–1673. [Google Scholar] [CrossRef]
- Sathesh-Prabu, C.; Kim, D.; Lee, K.S. Metabolic engineering of Escherichia coli for 2,3-butanediol production from cellulosic biomass by using glucose-inducible gene expression system. Bioresour. Technol. 2020, 309, 123361. [Google Scholar] [CrossRef] [PubMed]
- Mundhada, H.; Seoane, J.M.; Schneider, K.; Koza, A.; Christensen, H.B.; Klein, T.; Phaneuf, P.V.; Herrgard, M.; Feist, A.M.; Nielsen, A.T. Increased production of L-serine in Escherichia coli through Adaptive Laboratory Evolution. Metab. Eng. 2017, 39, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Rennig, M.; Mundhada, H.; Wordofa, G.G.; Gerngross, D.; Wulff, T.; Worberg, A.; Nielsen, A.T.; Norholm, M.H.H. Industrializing a Bacterial Strain for l-Serine Production through Translation Initiation Optimization. ACS Synth. Biol. 2019, 8, 2347–2358. [Google Scholar] [CrossRef] [PubMed]
- Bin, Z.; Liquan, R.; Miao, Y.; Ying, Z.; Ye, B. Enhanced l-ornithine production by systematic manipulation of l-ornithine metabolism in engineered Corynebacterium glutamicum S9114. Bioresour. Technol. 2018, 250, 60–68. [Google Scholar] [CrossRef]
- Datsenko KA, W.B. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, B.; Duan, C.; Sun, B.; Yang, J.; Yang, S. Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl. Environ. Microbiol. 2015, 81, 2506–2514. [Google Scholar] [CrossRef]
- Genshi, Z.; Malcolm, E.M. A Novel a-Ketoglutarate Reductase Activity of the serA-Encoded 3-Phosphoglycerate Dehydrogenase of Escherichia coli K-12 and Its Possible Implications for Human 2-Hydroxyglutaric Aciduria. J. Bacteriol. 1996, 178, 232–239. [Google Scholar]
- Schuller, D.J.; GrantZ, G.A.; Banaszak, L.J. The allosteric ligand site in the Vmax-type cooperative enzyme phosphoglycerate dehydrogenase. Struct. Biol. 1995, 2, 69–76. [Google Scholar] [CrossRef]
- Timothy, K.L.; Humgyu, H. Synthesis of l-tyrosine by a coupled reaction of serine hydroxymethyltransferase and β-tyrosinase. Enzym. Microb. Technol. 1986, 9, 523–526. [Google Scholar]
- Mei, H.; Dai, J.; Li, W.; Ling, X.; Zhu, P.; Zhao, Z. Comparison of Extraction Methods for E. coli Metabolome Analysis Using Liquid Chromatography Tandem Mass Spectrometry. Chin. J. Anal. Chem. 2011, 39, 534–539. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, Q.; Wei, G.; Liang, Q.; Qi, Q. Production in Escherichia coli of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) with differing monomer compositions from unrelated carbon sources. Appl. Environ. Microbiol. 2011, 77, 4886–4893. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Dujia, C.; Liang, W.; Juan, G.; Zhao, Q.; Wei, W.; Sun, Y. Comparative transcriptomic analysis reveals phenol tolerance mechanism of evolved Chlorella strain. Bioresour. Technol. 2017, 227, 266–272. [Google Scholar] [CrossRef]
- Grant, G.A.; Hu, Z.; Xu, X.L. Identification of Amino Acid Residues Contributing to the Mechanism of Cooperativity in Escherichia coli D-3-Phosphoglycerate Dehydrogenase. Biochemistry 2005, 44, 16844–16852. [Google Scholar] [CrossRef] [PubMed]
- Grant, G.A. D-3-Phosphoglycerate Dehydrogenase. Front. Mol. Biosci. 2018, 5, 110. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Hori, R.; Hemmi, H.; Downs, D.M.; Yoshimura, T. Inhibition of glycine cleavage system by pyridoxine 5′-phosphate causes synthetic lethality in glyA yggS and serA yggS in Escherichia coli. Mol. Microbiol. 2019, 113, 270–284. [Google Scholar] [CrossRef]
- Zhang, X.; Lai, L.; Xu, G.; Zhang, X.; Shi, J.; Koffas, M.A.G.; Xu, Z. Rewiring the Central Metabolic Pathway for High-Yield l-Serine Production in Corynebacterium glutamicum by Using Glucose. Biotechnol. J. 2019, 14, 1800497. [Google Scholar] [CrossRef]
- Ye, L.; Zhao, H.; Li, Z.; Wu, J.C. Improved acid tolerance of Lactobacillus pentosus by error-prone whole genome amplification. Bioresour. Technol. 2013, 135, 459–463. [Google Scholar] [CrossRef]
- Bhavani, S.; Trivedi, V.; Jala, V.R.; Subramanya, H.S.; Kaul, P.; Prakash, V.; Rao, N.A.; Savithri, H.S. Role of Lys-226 in the Catalytic Mechanism of Bacillus Stearothermophilus Serine Hydroxymethyltransferases Crystal Structure and Kinetic Studies. Biochemistry 2005, 44, 6929–6937. [Google Scholar] [CrossRef]
- Yu, L.; Gukui, C.; Xinwei, T.; Huitu, Z.; Xiaoguang, L.; Yihan, L.; Fuping, L. Construction of Escherichia coli strains producing L-serine from glucose. Biotechnol. Lett. 2012, 34, 1525–1530. [Google Scholar] [CrossRef]
- Burman, J.D.; Harris, R.L.; Hauton, K.A.; Lawson, D.M.; Sawers, R.G. The iron-sulfur cluster in the L-serine dehydratase TdcG from Escherichia coli is required for enzyme activity. FEBS Lett. 2004, 576, 442–444. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, X.M.; Wang, W.; Liu, Z.Q.; Zheng, Y.G. Metabolic engineering of Escherichia coli for d-pantothenic acid production. Food Chem. 2019, 294, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Cheng, Y.; Xie, X.; Xu, Q.; Chen, N. Modification of tryptophan transport system and its impact on production of L-tryptophan in Escherichia coli. Bioresour. Technol. 2012, 114, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wei, H.; Wang, T.; Xu, Q.; Zhang, C.; Fan, X.; Ma, Q.; Chen, N.; Xie, X. Current status on metabolic engineering for the production of l-aspartate family amino acids and derivatives. Bioresour. Technol. 2017, 245, 1588–1602. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, W.; Kim, Y.-M.; Mizushima, T.; Microbiology, D.O. Cloning and Expression of the Gene for the Na⫹-Coupled Serine Transporter from Escherichia coli and Characteristics of the Transporter. J. Bacteriol. 1998, 180, 6749–6752. [Google Scholar] [CrossRef]
- Schneider, F.; Kramer, R.; Burkovski, A. Identification and characterization of the main beta-alanine uptake system in Escherichia coli. Appl. Microbiol. Biotechnol. 2004, 65, 576–582. [Google Scholar] [CrossRef]
- Zhongqi, S.; Rongtuan, L.; Neman, E.B. Sequencing and characterization of the sdaC gene and identification of the sdaCB operon in Escherichia coli K12. Eur. J. Biochem. 1994, 222, 901–907. [Google Scholar]
- Sumantran, V.N.; Schweizer, H.P.; Datta, P. A Novel Membrane-Associated Threonine Permease Encoded by the tdcC Gene of Escherichia coli. J. Bacteriol. 1990, 172, 4288–4294. [Google Scholar] [CrossRef]
- Wang, C.; Wu, J.; Shi, B.; Shi, J.; Zhao, Z. Improving L-serine formation by Escherichia coli by reduced uptake of produced L-serine. Microb. Cell Factories 2020, 19, 66. [Google Scholar] [CrossRef]
- Simic, P.; Sahm, H.; Eggeling, L. L-Threonine Export: Use of Peptides to Identify a New Translocator from Corynebacterium glutamicum. J. Bacteriol. 2001, 183, 5317–5324. [Google Scholar] [CrossRef]
- Yen, M.; Tseng, Y.; Simic, P.; Sahm, H. The ubiquitous ThrE family of putative transmembrane amino acid efflux transporters. Res. Microbiol. 2002, 153, 19–25. [Google Scholar] [CrossRef]
- Fromm, H.J.; Hargrove, M. The Tricarboxylic Acid Cycle. In Essentials of Biochemistry; Springer: Berlin/Heidelberg, Germany, 2012; pp. 205–222. [Google Scholar]
- Hu, J.; Ma, H.; Zhang, W.; Yu, Z.; Sheng, G.; Fu, J. Effects of benzene and its metabolites on global DNA methylation in human normal hepatic l02 cells. Environ. Toxicol. 2014, 29, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Steinsiek, S.; Frixel, S.; Stagge, S.; Bettenbrock, K. Characterization of E. coli MG1655 and frdA and sdhC mutants at various aerobiosis levels. J. Biotechnol. 2011, 154, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shang, X.; Lai, S.; Zhang, Y.; Hu, Q.; Chai, X.; Wang, B.; Liu, S.; Wen, T. Reprogramming One-Carbon Metabolic Pathways to Decouple l-Serine Catabolism from Cell Growth in Corynebacterium glutamicum. ACS Synth. Biol. 2018, 7, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Chen, L.; Yang, Z.; Yao, L.; Zhu, J.; Qiu, J.; Wang, G.; He, J. The Properties of 5-Methyltetrahydrofolate Dehydrogenase (MetF1) and Its Role in the Tetrahydrofolate-Dependent Dicamba Demethylation System in Rhizorhabdus dicambivorans Ndbn-20. J. Bacteriol. 2019, 209, e00096-19. [Google Scholar] [CrossRef]
- Attia, Y.A.; Hassan, R.A.; Qota, E.M. Recovery from adverse effects of heat stress on slow-growing chicks in the tropics 1: Effect of ascorbic acid and different levels of betaine. Trop. Anim. Health Prod. 2009, 41, 807–818. [Google Scholar] [CrossRef]
- Yuewen, S.; Qunqun, G.; Wang, S.; Zhang, X.; Wang, J. Fed-Batch Fermentation of L-Threonine by Escherichia coli Supplemented with B-Vitamins. Adv. Biol. Sci. Res. 2017, 4, 285–291. [Google Scholar]
- De Lorenzo, V.; Sekowska, A.; Danchin, A. Chemical reactivity drives spatiotemporal organisation of bacterial metabolism. FEMS Microbiol. Rev. 2015, 39, 96–119. [Google Scholar] [CrossRef]
- Knapik, A.A.; Petkowski, J.J.; Otwinowski, Z.; Cymborowski, M.T.; Cooper, D.R.; Majorek, K.A.; Chruszcz, M.; Krajewska, W.M.; Minor, W. A multi-faceted analysis of RutD reveals a novel family of alpha/beta hydrolases. Proteins 2012, 80, 2359–2368. [Google Scholar] [CrossRef]
- Li, L.; Adachi, M.; Yu, J.; Kato, K.; Shinod, A.; Ostermann, A.; Schrader, T.E.; Ose, T.; Yao, M. Neutron crystallographic study of heterotrimeric glutamine amidotransferase CAB. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2019, 75, 193–196. [Google Scholar] [CrossRef]
- Zhang, Y.; Kang, P.; Liu, S.; Zhao, Y.; Wang, Z.; Chen, T. glyA gene knock-out in Escherichia coli enhances L-serine production without glycine addition. Biotechnol. Bioprocess Eng. 2017, 22, 390–396. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Description | Source |
|---|---|---|
| E.coli DH5α | Host of plasmid | Lab stock |
| E.coli BL21(DE3) | Expression strains | ATCC 26003 |
| E.coli W3110 | Parental strains | ATCC 23275 |
| SSW-01 | W3110 ΔsdaA | This work |
| SSW-02 | W3110 ΔsdaA glyA | This work |
| SSW-03 | W3110 ΔsdaA glyAmut | This work |
| SSW-05 | W3110 ΔsdaA glyAmut sdaB | This work |
| SSW-06 | W3110 ΔsdaA glyAmut ilvA | This work |
| SSW-07 | W3110 ΔsdaA glyAmut tdcB | This work |
| SSW-08 | W3110 ΔsdaA glyAmut tdcG | This work |
| SSW-10 | W3110 ΔsdaABC glyAmut | This work |
| Name | Description | Source |
|---|---|---|
| pT 7-7 | amp marker, T7 promoter | Takara |
| pSC | Low copy number, kan marker, p15A replicon, lambda PR and PL promoters | Lab stock |
| pKD13 | amp and kan markers | [16] |
| pKD46 | amp marker, temperature-sensitive | [16] |
| pCP20 | amp and chl makers, temperature-sensitive | [16] |
| pMD19-T simple | amp maker, TA cloning vector, 2692 bp | Takara |
| Pcas | repA101(Ts) kan Pcas-cas9 ParaB-Red lacIq Ptrc-sgRNA-pMB1 | [17] |
| PtargetF | pMB1 aadA | [17] |
| PtargetF-glyA | pMB1 aadA sgRNA-glyA | This work |
| pT-serA | pT 7-7 derivative, carrying serA | This work |
| pT-serAfr | pT 7-7 derivative, carrying serA | This work |
| pT-glyA | pT 7-7 derivative, carrying glyA | This work |
| pT-glyAmut | pT 7-7 derivative, carrying glyAmut | This work |
| SP-01 | SP derivative, carrying serAfr | This work |
| SP-02 | SP derivative, carrying serAfr, serC | This work |
| SP-05 | SP derivative, carrying serAfr, serC and serB | This work |
| SP-08 | SP derivative, carrying serAfr, serB, serC and pgk | This work |
| SP-09 | SP derivative, carrying serAfr, serB, serC, pgk and thrE | This work |
| Name | Sequence (5′–3′) a | Restriction Enzyme |
|---|---|---|
| serA-p1 | TCTAGAAAGAAGGAGATATACCATGGCAAAGGTATCGCTGGAG | Xba I |
| serA-p2 | GAGCTCGTGAGTAAGGGTAAGGGAGGATTG | Sac I |
| serB-p1 | AGATCTAAGAAGGAGATATACCATGCCTAACATTACCTGGTGCGACC | Bgl II |
| serB-p2 | AGTACTGGCTGATATCGGAGAGTTTCTGGAC | Sca I |
| serC-p1 | AGATCTAAGAAGGAGATATACCATGGCTCAAATCTTCAATTTTAG | Bgl II |
| serC-p2 | CAGCTGTACGATCGGCTGAAAGCGTATAG | pvu II |
| pgk-p1 | TCTAGAAAGAAGGAGATATACCATGTCTGTAATTAAGATGACCGATCTGG | Xba I |
| pgk-p2 | GAGCTCTTGATGGAGTCAGTACCGACG | Sac I |
| thrE-p1 | AGTACTAAGAAGGAGATATACCATGTTGAGTTTTGCGACCCTTCG | Sca I |
| thrE-p2 | AGATCTAGATCTACATCAAAGTGACGCCGTCGAAG | Xba I |
| Site directed mutagenesis primers used for mutation of serA | ||
| serA-p3 | GATGCACATCGCAGAAGCA | |
| serA-p4 | GCCCGGACGTGCTTCTGCGATGTGCATC | |
| glyA primers | ||
| glyA-F | GGAATTCCATATGTTAAAGCGTGAAATGAAC | Nde I |
| glyA-R | CCCAAGCTTTTATGCGTAAACCGGGTAAC | Hind III |
| sgRNA-F | TGGCAACCCACTTCAGCACCACTAGTATTATACCTAGGAC | |
| sgRNA-R | GGTGCTGAAGTGGGTTGCCAGTTTTAGAGCTAGAAATAGC | |
| glyA-D-F | TGTCCAACAGGACCGCCTATAAAGGCCAAAAATTTTATTGTTAGCTGAGTCAGGAGATGCGGATGTTAAAGCGTGAAATGAACATTG | |
| glyA-D-R | GGCGTTCACGCCGCATCCGGCATGAACAACGAGCACATTGACAGCAAATCACCGTTTCGCTTATGCGTAAACCGGGTAACG | |
| Position | Sequence Change | Protein Change | SHMT Activity (U) |
|---|---|---|---|
| -- | -- | -- | 0.65 ± 0.033 a |
| 24 | T → G | I8M (ATT → ATG) | 0.63 ± 0.022 |
| 88 | A → T | I30F (ATC → TTC) | 0.34 ± 0.005 |
| 145 | T → A | S49T (TCT → ACT) | 0.58 ± 0.005 |
| 149 | A → T | Q50L (CAG → CTG) | 0.24 ± 0.02 |
| 301 | G → T | A101S (GCT → TCT) | 0.22 ± 0.016 |
| 483 | A → T | Q161H (CAA → CAT) | 0.66 ± 0.018 |
| 532 | T → C | S178P (TCC → CCC) | 0.36 ± 0.028 |
| 572 | A → C | D191A (GAC → GCC) | 0.6 ± 0.019 |
| 685/686 | AA → GG | K229G (AAA → GGA) | 0.13 ± 0.005 |
| 743 | T → G | L248R (CTG → CGG) | 0.56 ± 0.014 |
| 902 | T → G | V301G (GTG → GGG) | 0.57 ± 0.017 |
| 1073 | T → C | V358A (GTG → GCG) | 0.54 ± 0.01 |
| 1163 | A → T | D388V (GAC → GTC) | 0.57 ± 0.008 |
| 1195 | A → T | I399F (ATC → TTC) | 0.62 ± 0.032 |
| 1225 | A → C | I409L (ATC → CTC) | 0.44 ± 0.004 |
| 1238 | A → C | Y413S (TAC → TCC) | 0.59 ± 0.012 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Li, Q.; Zhou, P.; Chen, X.; Shi, J.; Zhao, Z. Bioprocess Engineering, Transcriptome, and Intermediate Metabolite Analysis of L-Serine High-Yielding Escherichia coli W3110. Microorganisms 2022, 10, 1927. https://doi.org/10.3390/microorganisms10101927
Wang C, Li Q, Zhou P, Chen X, Shi J, Zhao Z. Bioprocess Engineering, Transcriptome, and Intermediate Metabolite Analysis of L-Serine High-Yielding Escherichia coli W3110. Microorganisms. 2022; 10(10):1927. https://doi.org/10.3390/microorganisms10101927
Chicago/Turabian StyleWang, Chenyang, Qinyu Li, Peng Zhou, Xiaojia Chen, Jiping Shi, and Zhijun Zhao. 2022. "Bioprocess Engineering, Transcriptome, and Intermediate Metabolite Analysis of L-Serine High-Yielding Escherichia coli W3110" Microorganisms 10, no. 10: 1927. https://doi.org/10.3390/microorganisms10101927
APA StyleWang, C., Li, Q., Zhou, P., Chen, X., Shi, J., & Zhao, Z. (2022). Bioprocess Engineering, Transcriptome, and Intermediate Metabolite Analysis of L-Serine High-Yielding Escherichia coli W3110. Microorganisms, 10(10), 1927. https://doi.org/10.3390/microorganisms10101927