SARS-CoV-2 ORF8 and SARS-CoV ORF8ab: Genomic Divergence and Functional Convergence

 , ,
, ,
Abstract
1. Introduction
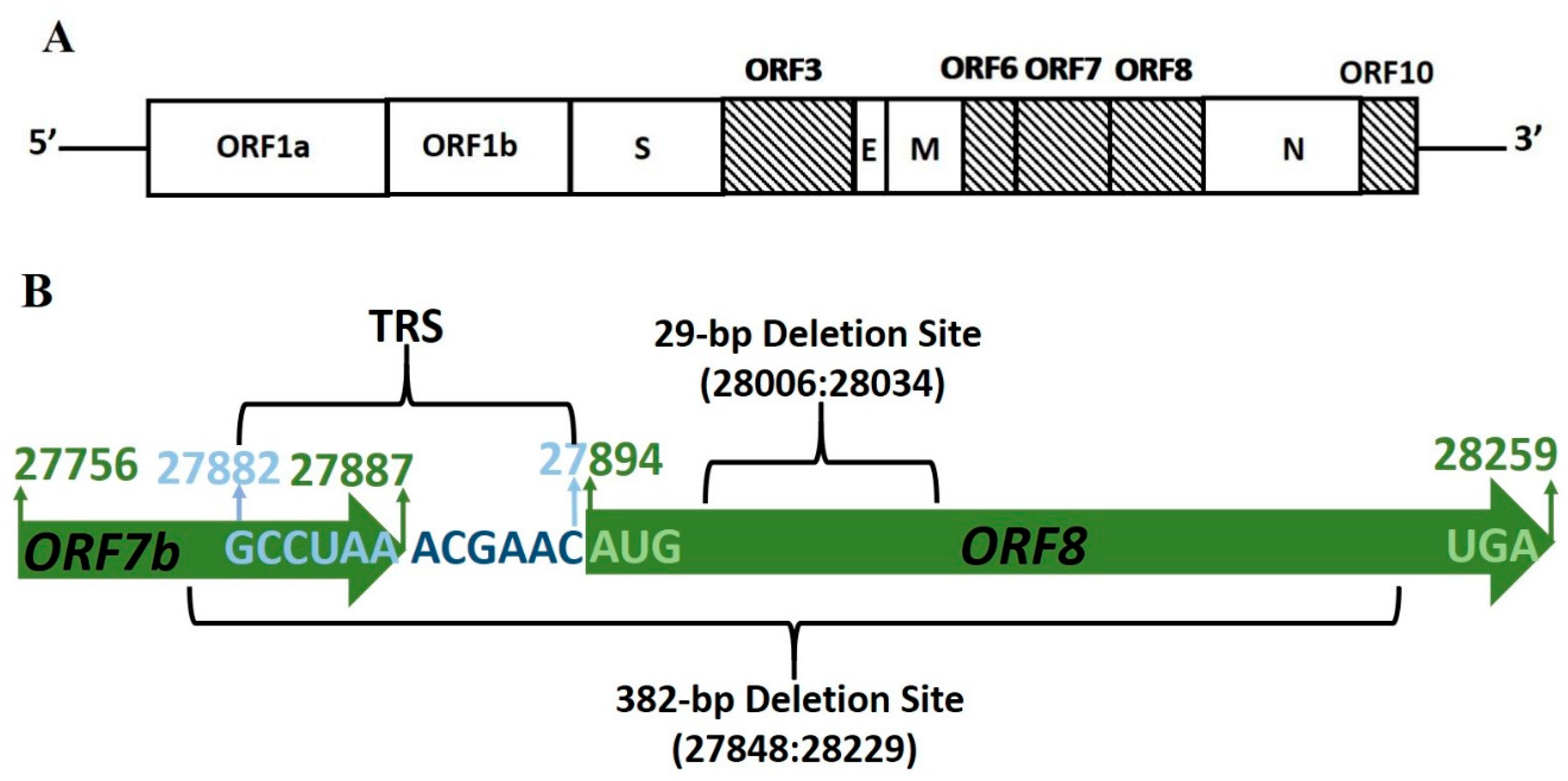
2. ORF8 Subgenomic mRNA8 Stability
2.1. Genome Deletions in SARS-CoV ORF8ab
2.2. Genome Deletions in SARS-CoV-2
2.3. Genomic Stability Estimation of SARS-CoV-2 ORF8
3. ORF8 Protein Origin
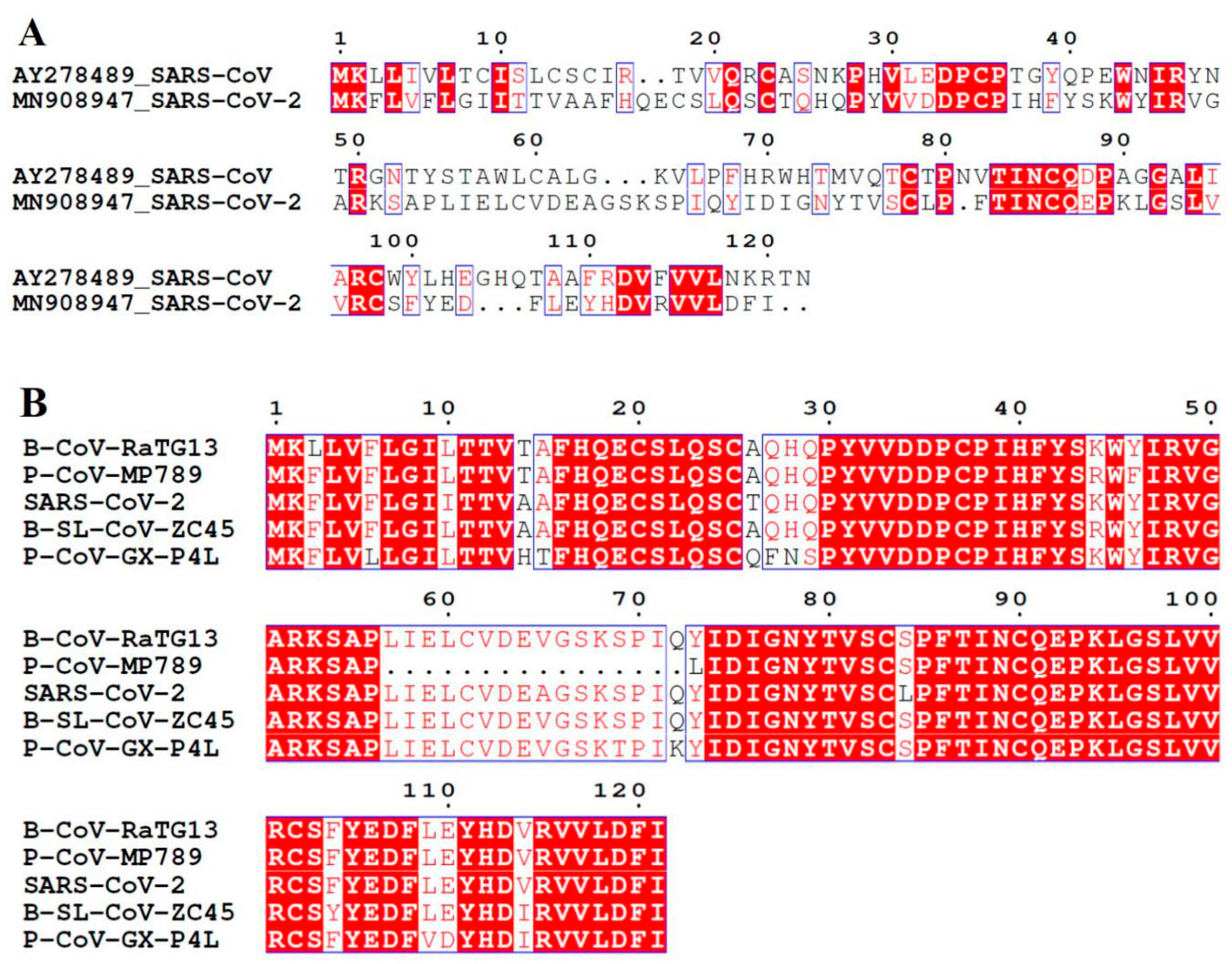
3.1. SARS-CoV ORF8ab Bat Origin
3.2. SARS-CoV-2 ORF8 Homologous Proteins
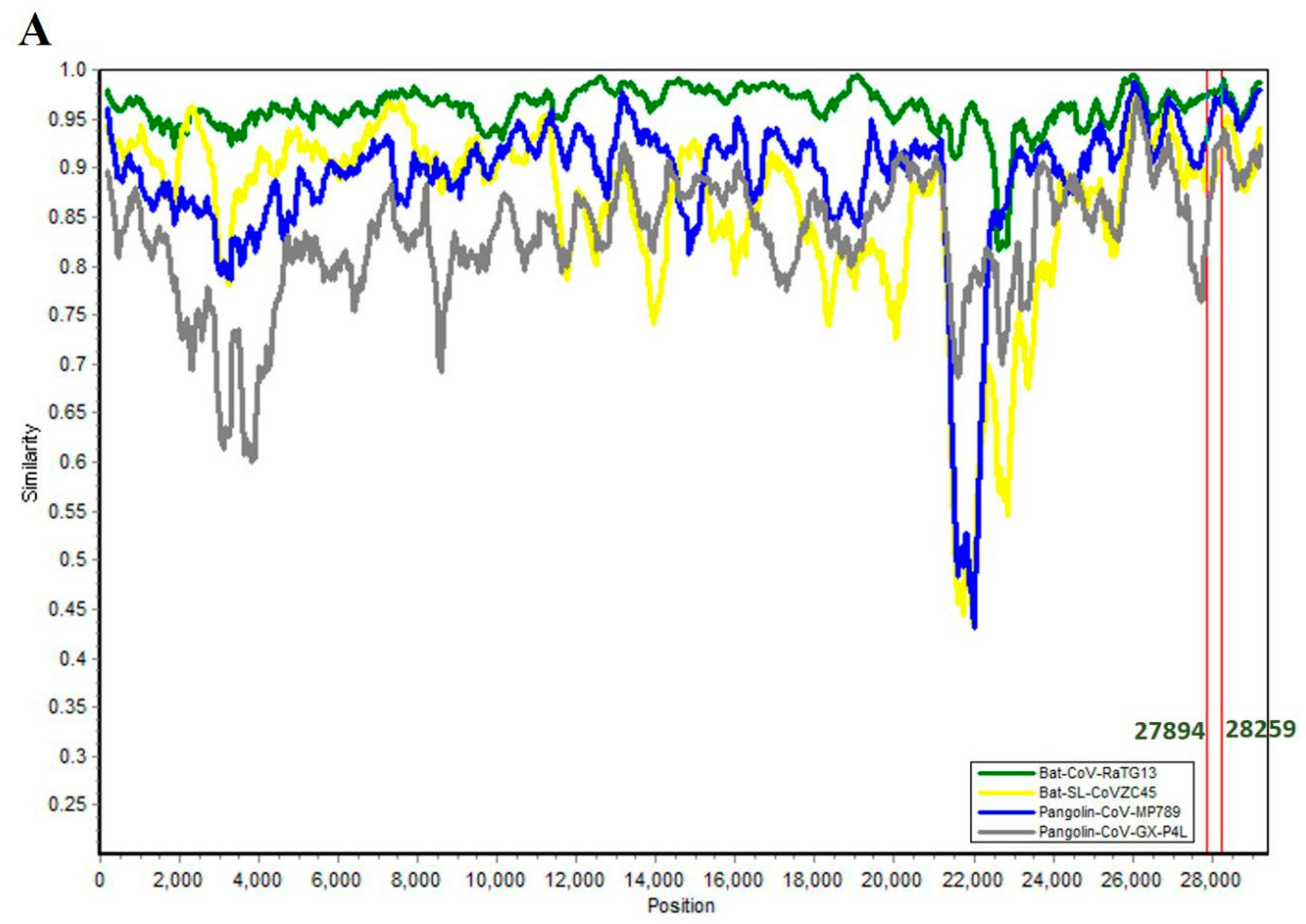
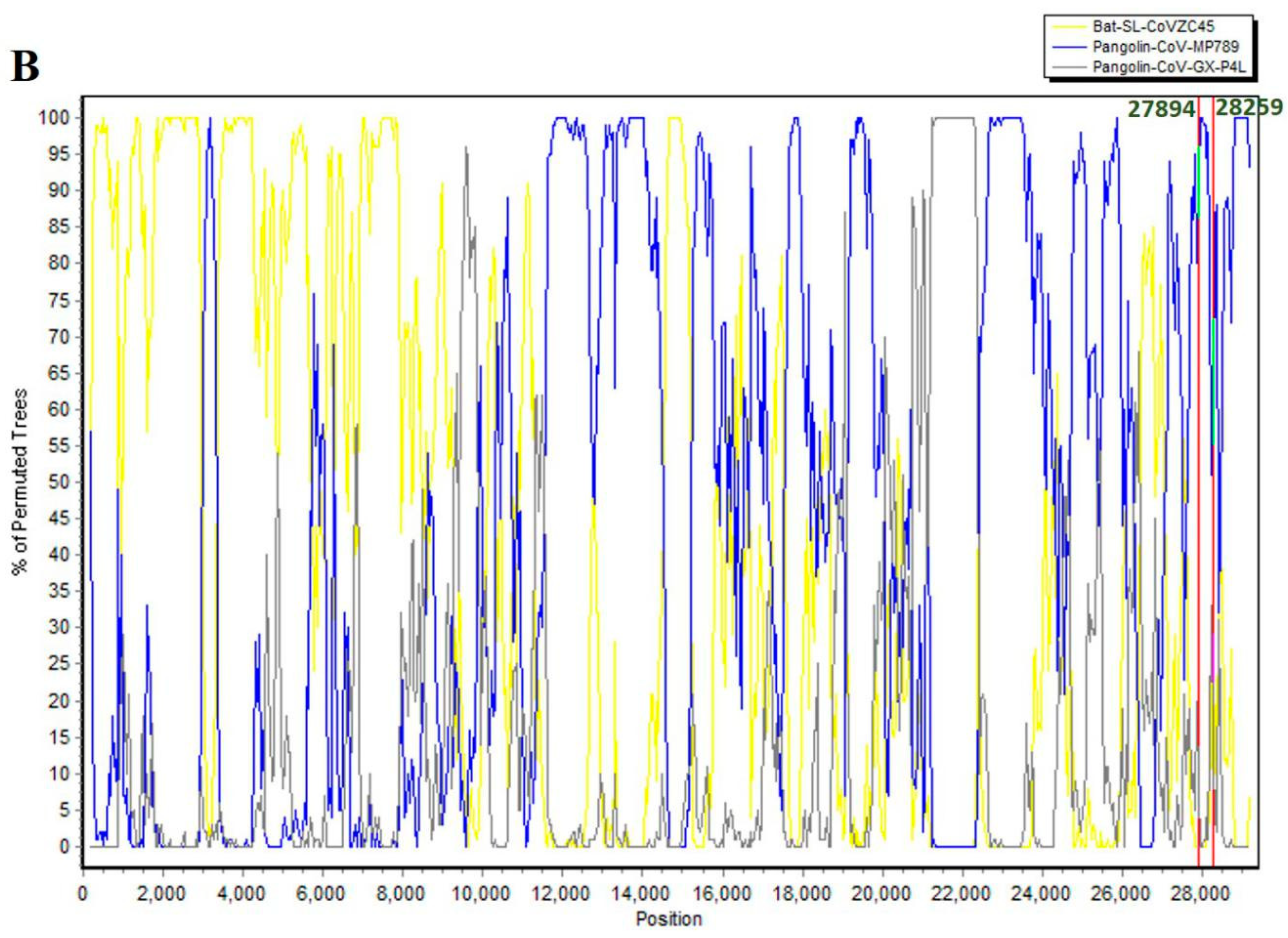
3.3. SARS-CoV-2 ORF8 Evolutionary Pathway
4. Conserved Features of SARS-CoV-2 ORF8 and SARS-CoV ORF8ab Proteins
4.1. Endoplasmic Reticulum Residence
4.2. Conversed Glycosylation Site
5. Functional Landscape of SARS-CoV-2 ORF8 and SARS-CoV ORF8ab
5.1. SARS-CoV ORF8ab, ORF8a and ORF8b Functions
5.1.1. Viral Replication
5.1.2. Immune Modulation
5.1.3. Unfolded Protein Response (UPR) Modulation
5.2. SARS-CoV-2 ORF8 Functions
5.2.1. Immune Modulation
5.2.2. Endoplasmic Reticulum Protein Quality Control
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome- related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [PubMed]
- WHO Director-General’s Opening Remarks at the Media Briefing on COVID-19—11 March 2020. Available online: https://www.who.int/dg/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020 (accessed on 22 July 2020).
- COVID-19 Dashboard by the Center for Systems Science and Engineering (CSSE) at Johns Hopkins University (JHU). Available online: https://coronavirus.jhu.edu/map.html (accessed on 23 July 2020).
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 3099, 19–20. [Google Scholar] [CrossRef]
- Wölfel, R.; Corman, V.M.; Guggemos, W.; Seilmaier, M.; Zange, S.; Müller, M.A.; Niemeyer, D.; Jones, T.C.; Vollmar, P.; Rothe, C.; et al. Virological assessment of hospitalized patients with COVID-2019. Nature 2020. [Google Scholar] [CrossRef]
- Liu, Y.; Yan, L.M.; Wan, L.; Xiang, T.X.; Le, A.; Liu, J.M.; Peiris, M.; Poon, L.L.M.; Zhang, W. Viral dynamics in mild and severe cases of COVID-19. Lancet Infect. Dis. 2020, 2019, 2019–2020. [Google Scholar] [CrossRef]
- Kissler, S.M.; Tedijanto, C.; Goldstein, E.; Yonatan, H.G.; Lipsitch, M. Projecting the transmission dynamics of SARS-CoV-2 through the postpandemic period. Science 2020, 21, 1–9. [Google Scholar] [CrossRef]
- Jia, J.S.; Lu, X.; Yuan, Y.; Xu, G.; Jia, J.; Christakis, N.A. Population flow drives spatio-temporal distribution of COVID-19 in China. Nature 2020, 1–11. [Google Scholar] [CrossRef]
- Bradley, B.T.; Maioli, H.; Johnston, R.; Chaudhry, I.; Fink, S.L.; Xu, H.; Najafian, B.; Marshall, D.; Lacy, J.M.; Williams, T.; et al. Histopathology and Ultrastructural Findings of Fatal COVID-19 Infections. medRxiv 2020. [Google Scholar] [CrossRef]
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary manifestations of COVID-19. Nat. Med. 2020, 26. [Google Scholar] [CrossRef]
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Cheng, L.; Li, J.; Wang, X.; Wang, F.; et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Sungnak, W.; Huang, N.; Bécavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R. Dysregulation of immune response in patients with COVID-19 in Wuhan, China Chuan. J. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Gu, J.; Han, B.; Wang, J. COVID-19: Gastrointestinal Manifestations and Potential Fecal–Oral Transmission. Gastroenterology 2020, 158, 1518–1519. [Google Scholar] [CrossRef] [PubMed]
- Endotypes, A.; Hariri, L.; Hardin, C.C. Covid-19, Angiogenesis and ARDS endotypes. N. Engl. J. Med. 2020, 1–2. [Google Scholar] [CrossRef]
- Galvan Casas, C.; Catala, A.; Carretero Hernandez, G.; Rodriguez-Jimenez, P.; Fernandez Nieto, D.; Rodriguez-Villa Lario, A.; Navarro Fernandez, I.; Ruiz-Villaverde, R.; Falkenhain, D.; Llamas Velasco, M.; et al. Classification of the cutaneous manifestations of COVID-19: A rapid prospective nationwide consensus study in Spain with 375 cases. Br. J. Dermatol. 2020. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; Kallio, K.; Kaya, T.; Anastasina, M.; Joensuu, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and provides a possible pathway into the central nervous system. bioRxiv 2020, 1–36. [Google Scholar] [CrossRef]
- Galeotti, C.; Bayry, J. Autoimmune and inflammatory diseases following COVID-19. Nat. Rev. Rheumatol. 2020. [Google Scholar] [CrossRef]
- Riphagen, S.; Gomez, X.; Gonzalez-Martinez, C.; Wilkinson, N.; Theocharis, P. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet 2020, 6736, 2019–2020. [Google Scholar] [CrossRef]
- Ace, H.; Prado, P.; Monteil, V.; Kwon, H.; Prado, P.; Hagelkru, A.; Wimmer, R.A.; Stahl, M.; Wirnsberger, G.; Zhang, H.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 3405, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature 2020. [Google Scholar] [CrossRef] [PubMed]
- Wanchao, Y.; Chunyou, M.; Xiaodong, L.; Dan-Dan, S.; Qingya, S.; Haixia, S.; Xiaoxi, W.; Fulai, Z.; Wenfeng, Z.; Minqi, G.; et al. Structural Basis for the Inhibition of the RNA-Dependent RNA Polymerase from SARS- CoV-2 by Remdesivir. Science 2020, 1560, 1–30. [Google Scholar]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 1–13. [Google Scholar] [CrossRef]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe Covid-19. N. Engl. J. Med. 2020, 1–13. [Google Scholar] [CrossRef]
- Zhang, X.; Tan, Y.; Ling, Y.; Lu, G.; Liu, F.; Yi, Z.; Jia, X.; Wu, M.; Wang, J.; Xu, M.; et al. Viral and host factors related to the clinical outcome of COVID-19. Nature 2020. [Google Scholar] [CrossRef]
- Li, G.; De Clercq, E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat. Rev. Drug Discov. 2020, 19, 149–150. [Google Scholar] [CrossRef]
- Narayanan, K.; Huang, C.; Makino, S. SARS coronavirus accessory proteins. Virus Res. 2008, 133, 113–121. [Google Scholar] [CrossRef]
- Liu, D.X.; Fung, T.S.; Chong, K.K.L.; Shukla, A.; Hilgenfeld, R. Accessory proteins of SARS-CoV and other coronaviruses. Antivir. Res. 2014, 109, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, L.; Geng, H.; Deng, Y.; Huang, B.; Guo, Y.; Zhao, Z.; Tan, W. The structural and accessory proteins M, ORF 4a, ORF 4b, and ORF 5 of Middle East respiratory syndrome coronavirus (MERS-CoV) are potent interferon antagonists. Protein Cell 2013, 4, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Eurosurveillance 2017, 22, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Salinas, L.; Zarate, S.; Eberl, S.; Gladue, D.P.; Novella, I.; Borca, M. V Positive selection of ORF3a and ORF8 genes drives the evolution of SARS-CoV-2 during the 2020 COVID-19 pandemic. bioRxiv 2020. [Google Scholar] [CrossRef]
- Oostra, M.; De Haan, C.A.M.; Rottier, P.J.M. The 29-Nucleotide Deletion Present in Human but Not in Animal Severe Acute Respiratory Syndrome Coronaviruses Disrupts the Functional Expression of Open Reading Frame 8. J. Virol. 2007, 81, 13876–13888. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, J.; Chen, Y.; Luo, B.; Yuan, Y.; Huang, F.; Yang, T.; Yu, F.; Liu, J.; Liu, B.; et al. The ORF8 Protein of SARS-CoV-2 Mediates Immune Evasion through Potently Downregulating MHC-I. bioRxiv 2020. [Google Scholar] [CrossRef]
- Li, J.-Y.; Liao, C.-H.; Wang, Q.; Tan, Y.-J.; Luo, R.; Qiu, Y.; Ge, X.-Y. The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus Res. 2020, 198074. [Google Scholar] [CrossRef]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 2003, 348, 1394–1398. [Google Scholar] [CrossRef]
- Guan, Y.; Zheng, B.J.; He, Y.Q.; Liu, X.L.; Zhuang, Z.X.; Cheung, C.L.; Luo, S.W.; Li, P.H.; Zhang, L.J.; Guan, Y.J.; et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in Southern China. Science 2003, 302, 276–278. [Google Scholar] [CrossRef]
- He, J.-F.; Peng, G.-W.; Min, J.; Yu, D.-W.; Liang, W.-J.; Zhang, S.-Y.; Xu, R.-H.; Zheng, H.-Y.; Wu, X.-W.; Xu, J.; et al. Molecular Evolution of the SARS Coronavirus during the Course of the SARS Epidemic in China. Science 2004, 303, 1666–1669. [Google Scholar]
- Koh, D.; Lim, M.; Ong, C.; Chia, S. Tracing SARS—Coronavirus Variant with Large Genomic Deletion. Emerg. Infect. Dis. 2005, 11, 168–170. [Google Scholar]
- Muth, D.; Corman, V.M.; Roth, H.; Binger, T.; Dijkman, R.; Gottula, L.T.; Gloza-Rausch, F.; Balboni, A.; Battilani, M.; Rihtarič, D.; et al. Attenuation of replication by a 29 nucleotide deletion in SARS-coronavirus acquired during the early stages of human-to-human transmission. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Holland, L.A.; Kaelin, E.A.; Maqsood, R.; Estifanos, B.; Wu, L.I.; Varsani, A.; Halden, R.U.; Hogue, B.G.; Scotch, M.; Lim, E.S. An 81 nucleotide deletion in SARS-CoV-2 ORF7a identified from sentinel surveillance in Arizona (Jan–Mar 2020). J. Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Anderson, D.E.; Young, B.E.; Zhu, F.; Linster, M.; Kalimuddin, S.; Low, J.; Yan, Z.; Jayakumar, J.; Sun, L.; et al. Discovery of a 382-nt deletion during the early evolution of SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Gong, Y.-N.; Tsao, K.-C.; Hsiao, M.-J.; Huang, C.-G.; Huang, P.-N.; Huang, P.-W.; Lee, K.-M.; Liu, Y.-C.; Yang, S.-L.; Kuo, R.-L.; et al. SARS-CoV-2 genomic surveillance in Taiwan revealed novel ORF8-deletion mutant and clade possibly associated with infections in Middle East. Emerg. Microbes Infect. 2020, 1–37. [Google Scholar] [CrossRef]
- Worobey, M.; Holmes, E.C. Evolutionary aspects of recombination in RNA viruses. J. Gen. Virol. 1999, 80, 2535–2543. [Google Scholar] [CrossRef]
- Hatcher, E.L.; Zhdanov, S.A.; Bao, Y.; Blinkova, O.; Nawrocki, E.P.; Ostapchuck, Y.; Schaffer, A.A.; Rodney Brister, J. Virus Variation Resource-improved response to emergent viral outbreaks. Nucleic Acids Res. 2017, 45, D482–D490. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Chen, S.; Zheng, X.; Zhu, J.; Ding, R.; Jin, Y.; Zhang, W.; Yang, H.; Zheng, Y.; Li, X.; Duan, G. Extended ORF8 Gene Region Is Valuable in the Epidemiological Investigation of Severe Acute Respiratory Syndrome—Similar Coronavirus. J. Infect. Dis. 2020, 222, 223–233. [Google Scholar] [CrossRef]
- Zwart, M.P.; Elena, S.F. Matters of Size: Genetic Bottlenecks in Virus Infection and Their Potential Impact on Evolution. Annu. Rev. Virol. 2015, 2, 161–179. [Google Scholar] [CrossRef]
- Mccrone, J.T.; Lauring, A.S. Genetic bottlenecks in intraspecies virus transmission. Curr. Opin. Virol. 2018, 28, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.B.; Swanstrom, R.; Kashuba, A.D.M.; Cohen, M.S. Bottlenecks in HIV—1 transmission: Insights from the study of founder viruses. Nat. Rev. Microbiol. 2015, 13, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Tsetsarkin, K.A.; Chen, R.; Leal, G.; Forrester, N.; Higgs, S.; Huang, J.; Weaver, S.C. Chikungunya virus emergence is constrained in Asia by lineage-specific adaptive landscapes. Proc. Natl. Acad. Sci. USA 2011, 108, 7872–7877. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, C.T.; McElhany, P.; Real, L.A. Transmission bottlenecks as determinants of virulence in rapidly evolving pathogens. Proc. Natl. Acad. Sci. USA 1999, 96, 5095–5100. [Google Scholar] [CrossRef] [PubMed]
- Hundertmark, K.J.; Daele, L.J. Van Founder effect and bottleneck signatures in an introduced, insular population of elk. Conserv. Genet. 2010, 11, 139–147. [Google Scholar] [CrossRef]
- Polymorphisms, H.I.V.; Bhattacharya, T.; Daniels, M.; Heckerman, D.; Foley, B.; Frahm, N.; Kadie, C.; Carlson, J.; Yusim, K.; Mcmahon, B.; et al. Founder Effects in the Assessment of HIV Polymorphisms and HLAAllele Associations. Science 2007, 315, 1583–1587. [Google Scholar]
- Farkas, C.; Fuentes-villalobos, F.; Garrido, J.L.; Jody, J. Insights on early mutational events in SARS-CoV-2 virus reveal founder effects across geographical regions. bioRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018, 46, D708–D717. [Google Scholar] [CrossRef] [PubMed]
- Azhar, E.I.; El-Kafrawy, S.A.; Farraj, S.A.; Hassan, A.M.; Al-Saeed, M.S.; Hashem, A.M.; Madani, T.A. Evidence for camel-to-human transmission of MERS coronavirus. N. Engl. J. Med. 2014, 370, 2499–2505. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.P.; Feng, Y.; Chen, H.; Luk, H.K.H.; Yang, W.-H.; Li, K.S.M.; Zhang, Y.-Z.; Huang, Y.; Song, Z.-Z.; Chow, W.-N.; et al. Severe Acute Respiratory Syndrome (SARS) Coronavirus ORF8 Protein Is Acquired from SARS-Related Coronavirus from Greater Horseshoe Bats through Recombination. J. Virol. 2015, 89, 10532–10547. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Lau, S.K.P.; Lam, C.S.F.; Lau, C.C.Y.; Tsang, A.K.L.; Lau, J.H.N.; Bai, R.; Teng, J.L.L.; Tsang, C.C.C.; Wang, M.; et al. Discovery of Seven Novel Mammalian and Avian Coronaviruses in the Genus Deltacoronavirus Supports Bat Coronaviruses as the Gene Source of Alphacoronavirus and Betacoronavirus and Avian Coronaviruses as the Gene Source of Gammacoronavirus and Deltacoronavi. J. Virol. 2012, 86, 3995–4008. [Google Scholar] [CrossRef] [PubMed]
- Latinne, A.; Hu, B.; Olival, K.J.; Zhu, G.; Zhang, L.; Li, H.; Chmura, A.A.; Field, H.E.; Zambrana-Torrelio, C.; Epstein, J.H.; et al. Origin and cross-species transmission of bat coronaviruses in China. bioRxiv 2020. [Google Scholar] [CrossRef]
- Banerjee, A.; Kulcsar, K.; Misra, V.; Frieman, M.; Mossman, K. Bats and coronaviruses. Viruses 2019, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.H.; McEachern, J.; Zhang, J.; Daszak, P.; Wang, H.; Field, H.; Li, W.; Eaton, B.T.; Wang, L.-F.; Yu, M.; et al. Bats Are Natural Reservoirs of SARS-Like Coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, L.; Ren, X.; Zhang, J.; Yang, F.; Zhang, S.; Jin, Q. ORF8-related genetic evidence for Chinese horseshoe bats as the source of human severe acute respiratory syndrome coronavirus. J. Infect. Dis. 2016, 213, 579–583. [Google Scholar] [CrossRef]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, 5–9. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Woo, P.C.Y.; Li, K.S.M.; Huang, Y.; Tsoi, H.W.; Wong, B.H.L.; Wong, S.S.Y.; Leung, S.Y.; Chan, K.H.; Yuen, K.Y. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. USA 2005, 102, 14040–14045. [Google Scholar] [CrossRef]
- Xiao, K.; Zhai, J.; Feng, Y.; Zhou, N.; Zhang, X.; Zou, J.-J.; Li, N.; Guo, Y.; Li, X.; Shen, X.; et al. Isolation and Characterization of 2019-nCoV-like Coronavirus from Malayan Pangolins. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zhang, T.; Wu, Q.; Zhang, Z. Pangolin homology associated with 2019-nCoV. bioRxiv 2020. [Google Scholar] [CrossRef]
- Liu, P.; Jiang, J.Z.; Wan, X.F.; Hua, Y.; Li, L.; Zhou, J.; Wang, X.; Hou, F.; Chen, J.; Zou, J.; et al. Are pangolins the intermediate host of the 2019 novel coronavirus (SARS-CoV-2)? PLoS Pathog. 2020, 16, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Wei, W.; Yuan, T.; Zheng, K.; Cui, X.; Li, J.; Pei, G.; Qiang, X.; Cheung, W.Y.; Li, L.; et al. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 2020, 583. [Google Scholar] [CrossRef]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Giorgi, E.E.; Marichannegowda, M.H.; Foley, B.; Xiao, C.; Kong, X.-P.; Chen, Y.; Gnanakaran, S.; Korber, B.; Gao, F. Emergence of SARS-CoV-2 through recombination and strong purifying selection. Sci. Adv. 2020, 6, eabb9153. [Google Scholar] [CrossRef]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-Length Human Immunodeficiency Virus Type 1 Genomes from Subtype C-Infected Seroconverters in India, with Evidence of Intersubtype Recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, 1–5. [Google Scholar] [CrossRef]
- Wong, M.C.; Cregeen, S.J.J.; Ajami, N.J.; Petrosino, J.F. Evidence of recombination in coronaviruses implicating pangolin origins of nCoV-2019. bioRxiv 2020, 2013. [Google Scholar] [CrossRef]
- Patino-Galindo, J.A.; Filip, I.; AlQuraishi, M.; Rabadan, R. Recombination and convergent evolution led to the emergence of 2019 Wuhan coronavirus. bioRxiv 2020, 1–13. [Google Scholar] [CrossRef]
- Xiang, D.; Shen, X.; Pu, Z.; Irwin, D.M.; Liao, M.; Shen, Y. Convergent evolution of human-isolated H7N9 avian influenza a viruses. J. Infect. Dis. 2018, 217, 1699–1707. [Google Scholar] [CrossRef]
- Zhang, C.; Mortuza, S.M.; He, B.; Wang, Y.; Zhang, Y. Template-based and free modeling of I-TASSER and QUARK pipelines using predicted contact maps in CASP12. Proteins 2018, 176, 139–148. [Google Scholar] [CrossRef]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Heo, L.; Feig, M. Modeling of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Proteins by Machine Learning and Physics-Based Refinement. bioRxiv 2020, 2. [Google Scholar] [CrossRef]
- Sung, S.; Chao, C.; Jeng, K.; Yang, J.; Lai, M.M.C. The 8ab protein of SARS-CoV is a luminal ER membrane-associated protein and induces the activation of ATF6. Virology 2009, 387, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.V.; Nagai, K.; Janda, C.Y.; Li, J.; Oubridge, C.; Herna, H. Recognition of a signal peptide by the signal recognition particle. Nature 2010, 465. [Google Scholar] [CrossRef]
- Signal Peptide Recognition. Nat. Rev. Mol. Cell Biol. 2010, 11, 2893. Available online: https://www.nature.com/articles/nrm2983 (accessed on 20 July 2020). [CrossRef]
- Martoglio, B. Signal sequences: More than just greasy peptides. Trends Cell Biol. 1998, 8, 14119–14123. [Google Scholar] [CrossRef]
- Feige, M.J.; Hendershot, L.M. Disulfide bonds in ER protein folding and homeostasis. Curr. Opin. Cell Biol. 2011, 23, 167–175. [Google Scholar] [CrossRef]
- Benham, A.M. Protein Secretion and the Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 2012, 4, a012872. [Google Scholar] [CrossRef]
- Young-Mi Go, D.P.J. Redox compartmentalization in eukaryotic cells. Biochim. Biophys. Acta 2008, 1780, 1273–1290. [Google Scholar] [CrossRef]
- Barlowe, C.K.; Miller, E.A. Secretory Protein Biogenesis and Traffic in the Early Secretory Pathway. Cell Struct. Traffick. 2013, 193, 383–410. [Google Scholar] [CrossRef]
- Ellgaard, L.; Sevier, C.S.; Bulleid, N.J. How Are Proteins Reduced in the Endoplasmic Reticulum? Trends Biochem. Sci. 2018, 43, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Le, T.M.; Wong, H.H.; Tay, F.P.L.; Fang, S.; Keng, C.T.; Tan, Y.J.; Liu, D.X. Expression, post-translational modification and biochemical characterization of proteins encoded by subgenomic mRNA8 of the severe acute respiratory syndrome coronavirus. FEBS J. 2007, 274, 4211–4222. [Google Scholar] [CrossRef] [PubMed]
- Vinh, P.; Lam, N.; Goldman, R.; Karagiannis, K.; Narsule, T.; Simonyan, V.; Soika, V.; Mazumder, R. Structure-based Comparative Analysis and Prediction of N-linked Glycosylation Sites in Evolutionarily Distant Eukaryotes. Genom. Proteomics Bioinform. 2013, 11, 96–104. [Google Scholar] [CrossRef]
- Xu, C.; Ng, D.T.W. Glycosylation-directed quality control of protein folding. Nat. Rev. Mol. Cell Biol. 2015, 16, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Li, C.; Wang, M.; Webb, G.I.; Zhang, Y.; Whisstock, J.C.; Song, J. GlycoMine: A machine learning-based approach for predicting N-, C- and O-linked glycosylation in the human proteome. Bioinformatics 2015, 31, 1411–1419. [Google Scholar] [CrossRef] [PubMed]
- Caragea, C.; Sinapov, J.; Silvescu, A.; Dobbs, D.; Honavar, V. Glycosylation site prediction using ensembles of Support Vector Machine classifiers. BMC Bioinform. 2007, 13, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bagdonaite, I.; Wandall, H.H. Global aspects of viral glycosylation. Glycobiology 2018, 28, 443–467. [Google Scholar] [CrossRef]
- Gupta, R.; Jung, E.; Brunak, S. Prediction of N-glycosylation sites in human proteins. Available online: http://www.cbs.dtu.dk/services/NetNGlyc/ (accessed on 20 July 2020).
- Watanabe, Y.; Bowden, T.A.; Wilson, I.A.; Crispin, M. BBA—General Subjects Exploitation of glycosylation in enveloped virus pathobiology. BBA—Gen. Subj. 2019, 1863, 1480–1497. [Google Scholar] [CrossRef]
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020, 9983, 1–9. [Google Scholar] [CrossRef]
- Chen, C.; Ping, Y.; Lee, H.; Chen, K.; Lee, Y.; Chan, Y.; Lien, T.; Jap, T.; Lin, C.; Kao, L.; et al. Open Reading Frame 8a of the Human Severe Acute Respiratory Syndrome Coronavirus Not Only Promotes Viral Replication but Also Induces Apoptosis. J. Infect. Dis. 2007, 112, 405–415. [Google Scholar] [CrossRef]
- Law, P.Y.P.; Liu, Y.M.; Geng, H.; Kwan, K.H.; Waye, M.M.Y.; Ho, Y.Y. Expression and functional characterization of the putative protein 8b of the severe acute respiratory syndrome-associated coronavirus. FEBS Lett. 2006, 580, 3643–3648. [Google Scholar] [CrossRef] [PubMed]
- Yount, B.; Roberts, R.S.; Sims, A.C.; Deming, D.; Frieman, M.B.; Sparks, J.; Denison, M.R.; Davis, N.; Baric, R.S. Severe Acute Respiratory Syndrome Coronavirus Group-Specific Open Reading Frames Encode Nonessential Functions for Replication in Cell Cultures and Mice. J. Virol. 2005, 79, 14909–14922. [Google Scholar] [CrossRef] [PubMed]
- Keng, C.T.; Åkerström, S.; Leung, C.S.W.; Poon, L.L.M.; Peiris, J.S.M.; Mirazimi, A.; Tan, Y.J. SARS coronavirus 8b reduces viral replication by down-regulating E via an ubiquitin-independent proteasome pathway. Microbes Infect. 2011, 13, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Keng, C.T.; Choi, Y.W.; Welkers, M.R.A.; Chan, D.Z.L.; Shen, S.; Gee Lim, S.; Hong, W.; Tan, Y.J. The human severe acute respiratory syndrome coronavirus (SARS-CoV) 8b protein is distinct from its counterpart in animal SARS-CoV and down-regulates the expression of the envelope protein in infected cells. Virology 2006, 354, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Totura, A.L.; Baric, R.S. SARS coronavirus pathogenesis: Host innate immune responses and viral antagonism of interferon. Curr. Opin. Virol. 2012, 2, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Hui, H.; Sing, T.; Fang, S.; Huang, M.; Tra, M.; Xiang, D. Accessory proteins 8b and 8ab of severe acute respiratory syndrome coronavirus suppress the interferon signaling pathway by mediating ubiquitin-dependent rapid degradation of interferon regulatory factor 3. Virology 2018, 515, 165–175. [Google Scholar]
- Luo, H. Interplay between the virus and the ubiquitin—Proteasome system: Molecular mechanism of viral pathogenesis. Curr. Opin. Virol. 2015, 17, 1–10. [Google Scholar] [CrossRef]
- Moshe, A.; Gorovits, R. Virus-Induced Aggregates in Infected Cells. Viruses 2012, 4, 2218–2232. [Google Scholar] [CrossRef]
- Shi, C.S.; Nabar, N.R.; Huang, N.N.; Kehrl, J.H. SARS-Coronavirus Open Reading Frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 2019, 5. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 in fl ammasome activation and its inhibitors. Cell Death Dis. 2019. [Google Scholar] [CrossRef]
- Kanneganti, T. Central roles of NLRs and inflammasomes in viral infection. Nat. Rev. Immunol. 2010, 10, 688–698. [Google Scholar] [CrossRef] [PubMed]
- He, B. Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ. 2006, 13, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Park, M.D. Immune evasion via SARS-CoV-2 ORF8 protein? Nat. Rev. Immunol. 2020, 111823. [Google Scholar] [CrossRef] [PubMed]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 6027. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, X.; Soloveva, V.; Warren, T.; Guo, F.; Wu, S.; Lu, H.; Guo, J.; Su, Q.; Shen, H.; et al. Enhancing the antiviral potency of ER α-glucosidase inhibitor IHVR-19029 against hemorrhagic fever viruses in vitro and in vivo. Antivir. Res. 2018, 150, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Warren, T.K.; Zhao, X.; Gill, T.; Guo, F.; Wang, L.; Ann, M.; Du, Y.; Alonzi, D.S.; Yu, W.; et al. Small molecule inhibitors of ER a -glucosidases are active against multiple hemorrhagic fever viruses. Antiviral Res. 2013, 98, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; You, Z.; Wang, Q.; Zhou, Z.; Qiu, Y.; Luo, R.; Ge, X. The epidemic of 2019-novel-coronavirus (2019-nCoV) pneumonia and insights for emerging infectious diseases in the future. Microbes Infect. 2020, 22, 80–85. [Google Scholar] [CrossRef]
- Hwang, J.; Qi, L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 2018, 43, 593–605. [Google Scholar] [CrossRef]
- Bernasconi, R.; Molinari, M. ERAD and ERAD tuning: Disposal of cargo and of ERAD regulators from the mammalian ER. Curr. Opin. Cell Biol. 2011, 23, 176–183. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ORF | Coordinates | Gene Length (nt) | CDS Length (nt) | TRS Location |
|---|---|---|---|---|
| 3a | 25,393–26,220 | 828 | 275 | 25,379 |
| 3b | 25,765–26,220 | 456 | 151 | |
| 6 | 27,202–27,387 | 186 | 61 | 27,035 |
| 7a | 27,394–27,759 | 366 | 121 | 27,382 |
| 7b | 27,756–27,887 | 132 | 43 | |
| 8 | 27,894–28,259 | 366 | 121 | 27,882 |
| 9a | 28,284–28,577 | 294 | 97 | |
| 9b | 28,734–28,955 | 222 | 73 | |
| 10 | 29,558–29,764 | 117 | 38 | 29,528 |
| Protein | Accession | Length [Coverage] | Percent Identity | Host | Isolate | Genome Accession | Genome Identity |
|---|---|---|---|---|---|---|---|
| Non-structural [NS8] | QHR63307.1 | 121[100] | 95.04 | Bat | CoV-Ra TG13 | MN996532 | 95.98 |
| Hypothetical | AVP78037.1 | 121[100] | 94.21 | Bat | SL-CoVZC45 | MG772933 | 84.69 |
| ORF8 | QIA48620.1 | 121[100] | 87.60 | Pangolin | PCoV-GX-P4L | MT040333.1 | 80.17 |
| ORF8 | QIG55952.1 | 105[100] | 81.82 | Pangolin | CoV-Isolate MP789 | MT121216.1 | 86.66 |
| Subject | Target | Genome Identity |
|---|---|---|
| SARS-CoV-2 | Bat-CoV-Ra TG13 | 95.98 |
| Bat-SL-CoVZC45 | 84.69 | |
| Pangolin-CoV-GX-789 | 86.66 | |
| Pangolin-CoV-GX-P4L | 80.17 | |
| Bat-CoV-Ra TG13 | Bat-SL-CoVZC45 Pangolin-CoV-MP789 | 85.3 89.3 |
| Pangolin-CoV-GX-P4L | 79.9 |
| No. | Begin | End | Recombinant Sequence(s) | Minor Parental Sequence(s) | Major Parental Sequence(s) | RDP p-Value |
|---|---|---|---|---|---|---|
| 1 | 927 | 1708 | SARS-CoV-2 | Bat-SL-CoVZC45 | Pangolin-CoV-MP789 | 2.00 × 10−6 |
| 2 | 1935 | 3194 | SARS-CoV-2 | Bat-SL-CoVZC45 | Pangolin-CoV-MP789 | 3.05 × 10−11 |
| 3 | 3664 | 4363 | SARS-CoV-2 | Bat-SL-CoVZC45 | Pangolin-CoV-MP789 | 3.84 × 10−9 |
| 4 | 22,874 | 23,092 | SARS-CoV-2 | Pangolin-CoV-GX-P4L | Bat-SL-CoVZC45 | 2.52 × 10−2 |
| 5 | 23,156 | 23,306 | SARS-CoV-2 | Pangolin-CoV-GX-P4L | Bat-SL-CoVZC45 | 5.09 × 10−3 |
| 6 | 23,898 | 24,248 | Bat-CoV-RaTG13 | Pangolin-CoV-GX-P4L | Bat-SL-CoVZC45 | 1.47 × 10−3 |
| SARS-CoV-2 | ||||||
| 7 | 6649 | 6833 | Bat-CoV-RaTG13 | Bat-SL-CoVZC45 | Pangolin-CoV-MP789 | 3.19 × 10−2 |
| SARS-CoV-2 |
| Number | Begin | End | Recombinant Sequence(s) | Minor Parental Sequence(s) | Major Parental Sequence(s) | RDP p-Value |
|---|---|---|---|---|---|---|
| 1 | 380 | 11,623 | Pangolin-CoV-GX-P4L | Unknown (Bat-SL-CoVZC45) | SARS-CoV-2 | 1.27 × 10−57 |
| 2 | 7054 | 8258 | Pangolin-CoV-MP789 | Unknown (Bat-SL-CoVZC45) | SARS-CoV-2 | 1.36 × 10−10 |
| 3 | 9558 | 9947 | Pangolin-CoV-MP789 | Bat-SL-CoVZC45 | SARS-CoV-2 | 2.20 × 10−2 |
| 4 | 14,611 | 15,451 | Pangolin-CoV-MP789 | Unknown (Bat-SL-CoVZC45) | SARS-CoV-2 | 8.00 × 10−13 |
| 5 | 17,813 | 18,698 | Bat-SL-CoVZC45 | Unknown (Pangolin-CoV-GX-P4L) | SARS-CoV-2 | 9.99 × 10−3 |
| 6 | 19,847 | 19,963 | Pangolin-CoV-MP789 | Pangolin-CoV-GX-P4L | SARS-CoV-2 | 5.09 × 10−9 |
| 7 | 21,563 | 21,904 | Pangolin-CoV-MP789 | Unknown (Pangolin-CoV-GX-P4L) | SARS-CoV-2 | 1.34 × 10−9 |
| 8 | 21,914 | 22,474 | Pangolin-CoV-MP789 | Unknown (Pangolin-CoV-GX-P4L) | SARS-CoV-2 | 3.73 × 10−21 |
| 9 | 22,850 | 23,094 | Bat-CoV-RaTG13 | Unknown (Pangolin-CoV-MP789) | SARS-CoV-2 | 1.70 × 10−16 |
| 10 | 4816 | 5953 | Pangolin-CoV-MP789 | Unknown (Bat-SL-CoVZC45) | Bat-CoV-RaTG13 | 1.25 × 10−9 |
| SARS-CoV-2 | ||||||
| 11 | 14,042 | 14,607 | Bat-SL-CoVZC45 | Unknown (Pangolin-CoV-GX-P4L) | Pangolin-CoV-MP789 | 2.13 × 10−4 |
| SARS-CoV-2 | ||||||
| Bat-CoV-RaTG13 | ||||||
| 12 | 16,028 | 16,399 | Bat-SL-CoVZC45 | Unknown (Pangolin-CoV-GX-P4L) | Bat-CoV-RaTG13 | 2.07 × 10−4 |
| SARS-CoV-2 | ||||||
| 13 | 21,187 | 22,368 | Bat-SL-CoVZC45 | Pangolin-CoV-MP789 | Bat-CoV-RaTG13 | 1.69 × 10−45 |
| SARS-CoV-2 | ||||||
| 14 | 20,015 | 20,591 | Bat-SL-CoVZC45 | Unknown (Pangolin-CoV-GX-P4L) | Bat-CoV-RaTG13 | 1.42 × 10−7 |
| SARS-CoV-2 | ||||||
| 15 | 22,472 | 22,792 | Pangolin-CoV-GX-P4L | Bat-CoV-RaTG13 | Unknown (Bat-SL-CoVZC45) | 3.91 × 10−4 |
| SARS-CoV-2 |
| Characteristic | SARS-CoV ORF8ab | SARS-COV-2 ORF8 |
|---|---|---|
| Nucleotide Identity | 26% | |
| Protein Identity | 20% | |
| Nucleotide Deletion | Yes | No (This Study) |
| Origin | Bat | Bat, Pangolin (This Study) |
| Biochemical/Structural Features | ||
| N-Terminal Peptide Sequence | Yes | Yes (Predicted in this Study) |
| N-Glycosylation Site | Yes | Yes (Identified in this Study) |
| Cysteine Residues | Yes | Yes |
| Localization | Endoplasmic Reticulum | Endoplasmic Reticulum (Predicted/proposed in this Study) |
| Protein Family Conserved Motifs | Yes | Yes |
| Macromolecular Interactions | Protein–protein | Protein–protein and Protein–DNA |
| Functional Features | ||
| Viral Replication | Yes | Not studied so far |
| Host Immune Modulation | Yes | Yes |
| Protein Quality Control | Yes | Yes |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammad, S.; Bouchama, A.; Mohammad Alharbi, B.; Rashid, M.; Saleem Khatlani, T.; Gaber, N.S.; Malik, S.S. SARS-CoV-2 ORF8 and SARS-CoV ORF8ab: Genomic Divergence and Functional Convergence. Pathogens 2020, 9, 677. https://doi.org/10.3390/pathogens9090677
Mohammad S, Bouchama A, Mohammad Alharbi B, Rashid M, Saleem Khatlani T, Gaber NS, Malik SS. SARS-CoV-2 ORF8 and SARS-CoV ORF8ab: Genomic Divergence and Functional Convergence. Pathogens. 2020; 9(9):677. https://doi.org/10.3390/pathogens9090677
Chicago/Turabian StyleMohammad, Sameer, Abderrezak Bouchama, Bothina Mohammad Alharbi, Mamoon Rashid, Tanveer Saleem Khatlani, Nusaibah S. Gaber, and Shuja Shafi Malik. 2020. "SARS-CoV-2 ORF8 and SARS-CoV ORF8ab: Genomic Divergence and Functional Convergence" Pathogens 9, no. 9: 677. https://doi.org/10.3390/pathogens9090677
APA StyleMohammad, S., Bouchama, A., Mohammad Alharbi, B., Rashid, M., Saleem Khatlani, T., Gaber, N. S., & Malik, S. S. (2020). SARS-CoV-2 ORF8 and SARS-CoV ORF8ab: Genomic Divergence and Functional Convergence. Pathogens, 9(9), 677. https://doi.org/10.3390/pathogens9090677