1. Introduction
The innate immune response (IIR) is the first line of defense against infection, which involves the activation of different pathways through the recognition of pathogen-associated molecular patterns (PAMPs) by different pattern recognition receptors (PRRs). This response is strongly regulated at different levels, including the epigenetic mechanisms.
Mammary gland epithelial cells are targets of different reproductive hormones, such as prolactin (PRL) and estradiol (E2). These hormones regulate the proliferation, differentiation and survival of the mammary epithelium. Additionally, they can modulate the IIR of this tissue; however, the epigenetic role of these hormones in this response is little known [
1,
2]. In this sense, PRL and E2 inflammatory functions are diverse in several physiologic and pathologic conditions [
3,
4]. In mammary glands, these hormones can act alone or in combination, regulating the immune microenvironment [
5]. In bovines, during the period around parturition, there is an increased susceptibility to inflammatory disorders in the mammary gland, such as mastitis [
6]. This susceptibility has been correlated with abrupt hormonal changes, for example, E2 levels rise suddenly in the last week before parturition, increase in the last 3 days before delivery and fall rapidly after calving at basal values [
6,
7]. On the other hand, PRL also increases at calving, triggering lactogenesis and galactopoeisis [
8]. In addition, E2 can potentiate the effects of others hormones on the mammary epithelium, for example, E2 with PRL stimulate proliferation more than either hormone alone and promote the progression of the epithelial structures toward a more differentiated state [
9,
10].
Bovine mammary epithelial cells (bMECs) play a relevant role during intramammary infections because they are in close contact with the bacteria responsible of mastitis, becoming a target for persistent bacteria causing chronic and subclinical infections, such as
Staphylococcus aureus [
11,
12]. In addition, bMECs are able to orchestrate a relevant defense against infection [
13]. The role of bovine PRL (bPRL) and E2 in the susceptibility of bMECs during
S. aureus infection has been explored in our group evaluating either hormone alone, showing that bPRL at physiological concentrations (5 ng/mL) induces the internalization of
S. aureus into bovine mammary epithelial cells, whereas E2 (50 pg/mL) reduces it. Both effects are achieved through the modulation of elements of the IIR of bMECs, such as cytokines and antimicrobial peptide production [
14,
15]. However, we do not know if the combination of these hormones, resembling an in vivo condition, could improve the defense response of bMECs. In addition, the epigenetic modulation of bMECs during
S. aureus infection has been explored both in vitro as well in vivo. In this sense, Modak et al. [
16], have reported that in a mice model for
S. aureus-induced mastitis, bacteria induced hyperacetylation at histone H3K9 and H3K14 residues in mammary tissue. These acetylation marks were enriched at the promoters of overexpressed proinflammatory genes. Furthermore, Kweh et al. [
17], have showed that the expression of β-defensins in bMECs is under the epigenetic control of DNA methylation and histone acetylation. In addition, the inhibition of histone deacetylases (HDACs) in bMECs inhibits the expression of inflammatory genes [
18].
In spite of the evidence indicating an immunomodulatory role of hormones during S. aureus infection in bMECs, as well as the evidence showing the epigenetic regulation of inflammatory genes during mastitis, there are no studies related to the epigenetic hormonal modulation of the mammary epithelium during infection. Thus, the objective of this work was to analyze the effects of combined bPRL and E2 on bMEC defense during S. aureus infection and to determine if they can induce epigenetic modifications.
3. Discussion
Cattle are prone to develop intramammary infections (IMIs) during the lactating cycle of the mammary gland. Although a higher susceptibility to IMIs occurs during the transition period (period from late pregnancy until early lactation), which is commonly defined as the period from 3 weeks before to 3 weeks after calving, IMIs can be also detected in non-lactating dairy cows [
20,
21]. This susceptibility has been correlated with hormonal changes, for example, E2 levels rise abruptly in the last week before parturition, and bPRL also increases at calving, triggering lactation [
8]. In our group, we have previously explored the role of bPRL and E2 (separately) on the IIR of the bMECs, the target cells of these hormones and the host of intracellular
S. aureus. bPRL induces
S. aureus internalization into bMECs [
19], but E2 reduces it [
15], with both effects at physiological concentrations of the hormones. Since both hormones act together on bMECs in vivo, in this work, we explore the regulation of the elements of the IIR of these cells by the combination of hormones bPRL and E2 during
S. aureus infection. Furthermore, we analyze if these effects could be related to epigenetic modulation.
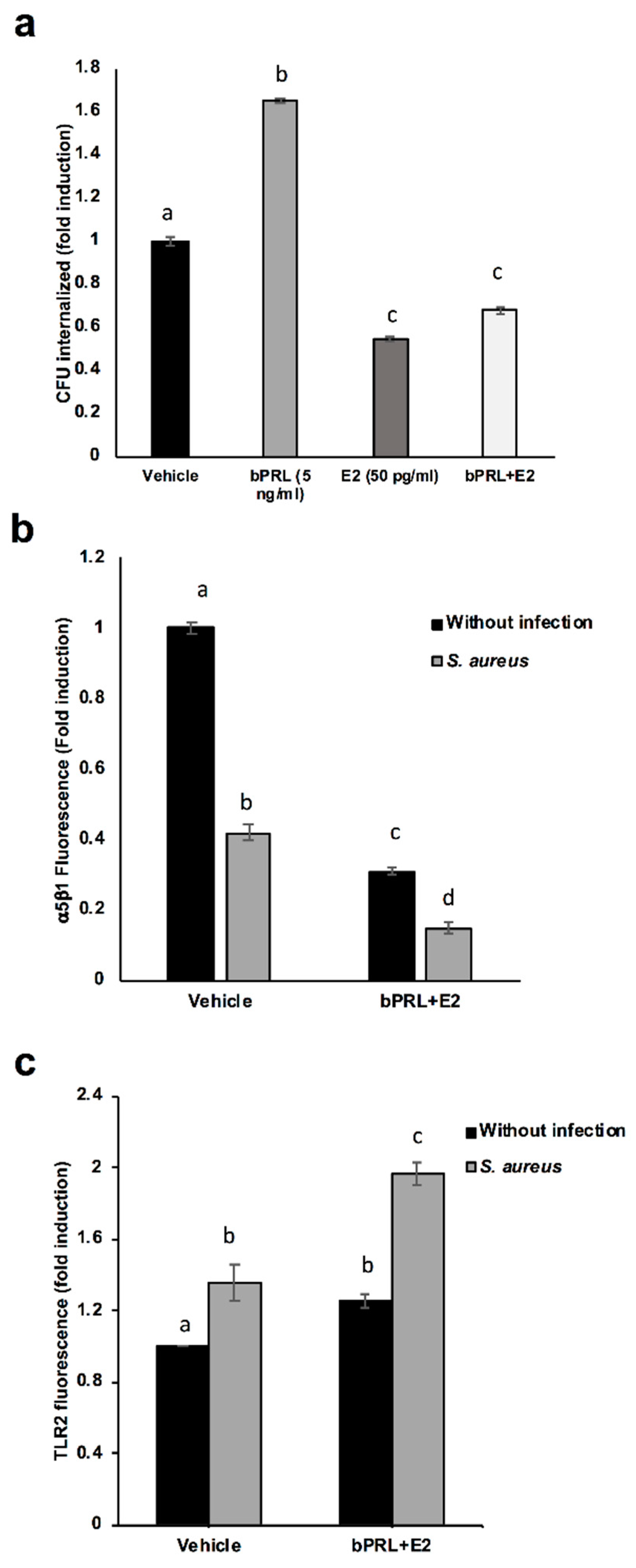
First, we determined that the combined hormones neither significantly affected the viability of bacteria nor altered bMEC proliferation. These results agree with previous reports from our group [
14,
15]. On the other hand, combined hormones reduced
S. aureus internalization into bMECs (35%), in a similar way to E2 [
15]. In order to explain this reduction, the α5β1 integrin MA was evaluated, as it is the host receptor during
S. aureus internalization in different epithelial cells. The α5β1 MA of bMECs was reduced by 70% in cells treated for 24 h with combined hormones (
Figure 2b). The reduction in the MA observed in this receptor upon infection correlates with a previous report from our group, indicating that the internalization of bacteria results in the endocytosis of this receptor [
19]. In this work, the α5β1 integrin MA was evaluated in cells infected for 2 h; however, we previously determined a reduction in the MA of this receptor after 15 min of infection [
19]. Thus, the reduction in bacterial internalization could be related to the reduced MA of integrin α5β1 in hormone pre-treated bMECs.
With the purpose of determining if the combined hormones modulate the IIR of bMECs, the MA of TLR2 was analyzed, as this receptor is one of the main innate immune receptors recognizing
S. aureus [
22]. We have demonstrated that
S. aureus induces TLR2 MA and activation in bMECs, and with respect to the hormones, bPRL stimulates TRL2 MA, whereas E2 reduces it [
15,
19]. In this work, combined hormones increased TLR2 MA (~25%), which was enhanced by infection (~100%). This result indicates that the combined hormones could favor the activation of the IIR of bMECs. Similar reports for the combination of bPRL and E2 have not been described in mammary glands; however, the role of these hormones in the immune microenvironment of mammary glands has been proposed [
5]. In agreement with the activation of TLR2 induced by the combined hormones, we detected that the cytokine profile of bMECs stimulated by bPRL and E2 is mainly pro-inflammatory: hormones induce the expression of TNF-α and IL-1β, but also induce the expression of IL-10. This profile is maintained during infection, with the exception of IL-β, which is reduced in the presence of
S. aureus. In addition, hormones up-regulate the expression of CXCL8 in infected bMECs. This effect is different from that reported previously for bPRL and E2 added separately to bMECs [
15,
19]. In particular, the up-regulation in CXCL8 expression is a remarkable effect of the combined hormones during infection, and it would be interesting to analyze in further research if leukocyte chemotaxis is induced under these conditions. According to previous work, the cytokine gene expression correlates with protein production [
15]; thus, it is necessary in future experiments to perform protein determination in order to corroborate this. In this work, we also demonstrate that the combined hormones induce the expression of different antimicrobial peptides, which is maintained after infection. This effect was not detected with hormones added separately, indicating that the hormonal combination could lead to a better defense against
S. aureus. A synergism achieved by PRL and E2 on the expression of different genes has been reported in human mammary cells [
23], but the cross-talking between PRL and E2 signaling pathways requires further research.
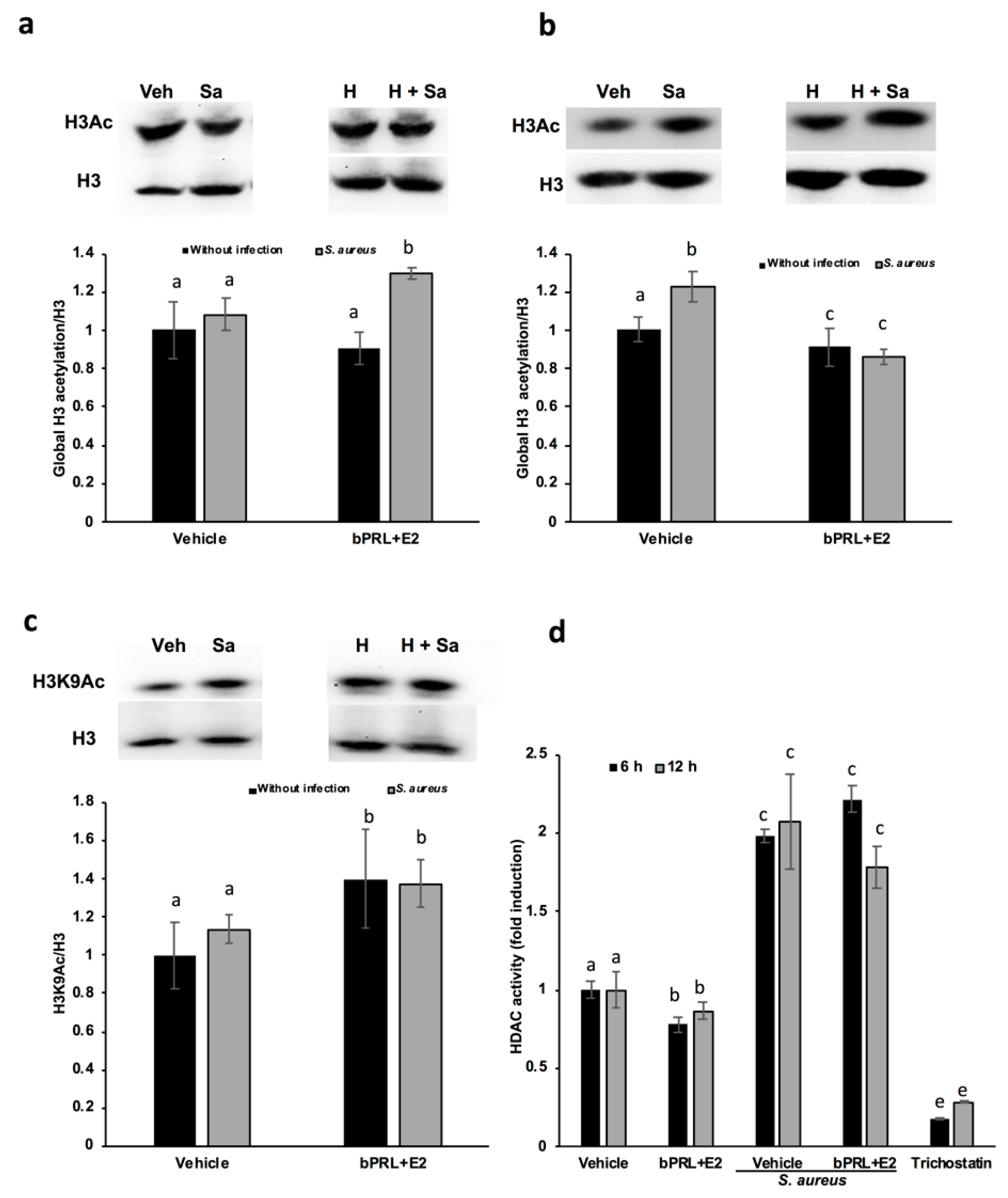
In this work, we attempt to correlate the effect of hormones on innate immune genes with epigenetic marks. The analysis of the epigenetic modulation was carried out for different durations (6, 12 and 24 h) of hormonal stimulation, because the epigenetic effects on chromatin can be achieved with short durations [
16], and the effects detected on gene expression at 24 h could be the consequence of immediate chromatin rearrangement. In this work, we analyzed post-translational modifications in histone H3K9, such as acetylation and dimethylation; additionally, the role of HDACs and HDMs in the abovementioned epigenetic marks was evaluated. Our results indicate that the global acetylation of H3 increases in bMECs infected and treated for 12 h with the combined hormones. This mark was down-regulated at 24 h of hormonal treatment (
Figure 3). The H3K9Ac could be related to the global H3Ac, considering that we detected an up-regulation in this mark at 12 h of hormonal treatment in bMECs before infection, but interestingly, this mark was maintained after 2 h of infection with
S. aureus. In agreement, Modak et al. (2014) reported that this mark is induced by
S. aureus in mouse mammary glands [
16], using an in vivo mastitis model. In our model, we detected a subtle up-regulation in H3K9Ac induced by
S. aureus, which was enhanced by the combined hormones. To our knowledge, this is the first report that evaluates the H3K9Ac mark in the bovine mammary epithelium in response to bPRL and E2, which could be related to the up-regulation detected in the inflammatory genes. In addition, this mark can be also related to the global H3Ac, because the antibody employed detects the acetylation in lysine residues K9, K14, K18, K23 and K27. In this sense, it is necessary to analyze the other marks, given that H3K14ac is up-regulated in mouse mammary glands upon
S. aureus infection [
16]. The H3K9Ac mark induced by the combined hormones at 12 h of treatment coincides with the reduction in the activity of HDACs at 6 and 12 h (
Figure 4d). This reduction indicates that the histone acetylation is maintained because the enzymes that remove acetylation marks are down-regulated or inhibited. Accordingly, Kweh et al. (2019) have reported that HDACs participate in the regulation of the expression of antimicrobial peptides in primary bMECs [
17]. In addition, Romanick et al. (2018) indicate that HDACs 1 and 2 regulate inflammatory gene expression via canonical and noncanonical mechanisms in the bovine mammary epithelial cell line MAC-T [
18]. In this work, we also demonstrated that
S. aureus induces HDAC activation in bMECs. To our knowledge, this is the first report that shows this activity induced by
S. aureus. In addition, Silva et al. (2018) have shown that short chain fatty acids inhibited HDAC activity, whereas they increased H3Ac in MAC-T cells [
24]. We observed a similar effect in this work with the combined hormones. The kit employed to analyze HDACs detects the activity of class I HDACs (HDAC1, 2, 3 and 8), IIa (HDAC4, 5, 7 and 9), and IIb (HDAC6 and 10). We did not detect any change in the levels of HDAC1 gene expression (data not shown), therefore other HDAC(s) should be implicated in the reduced enzymatic activity observed.
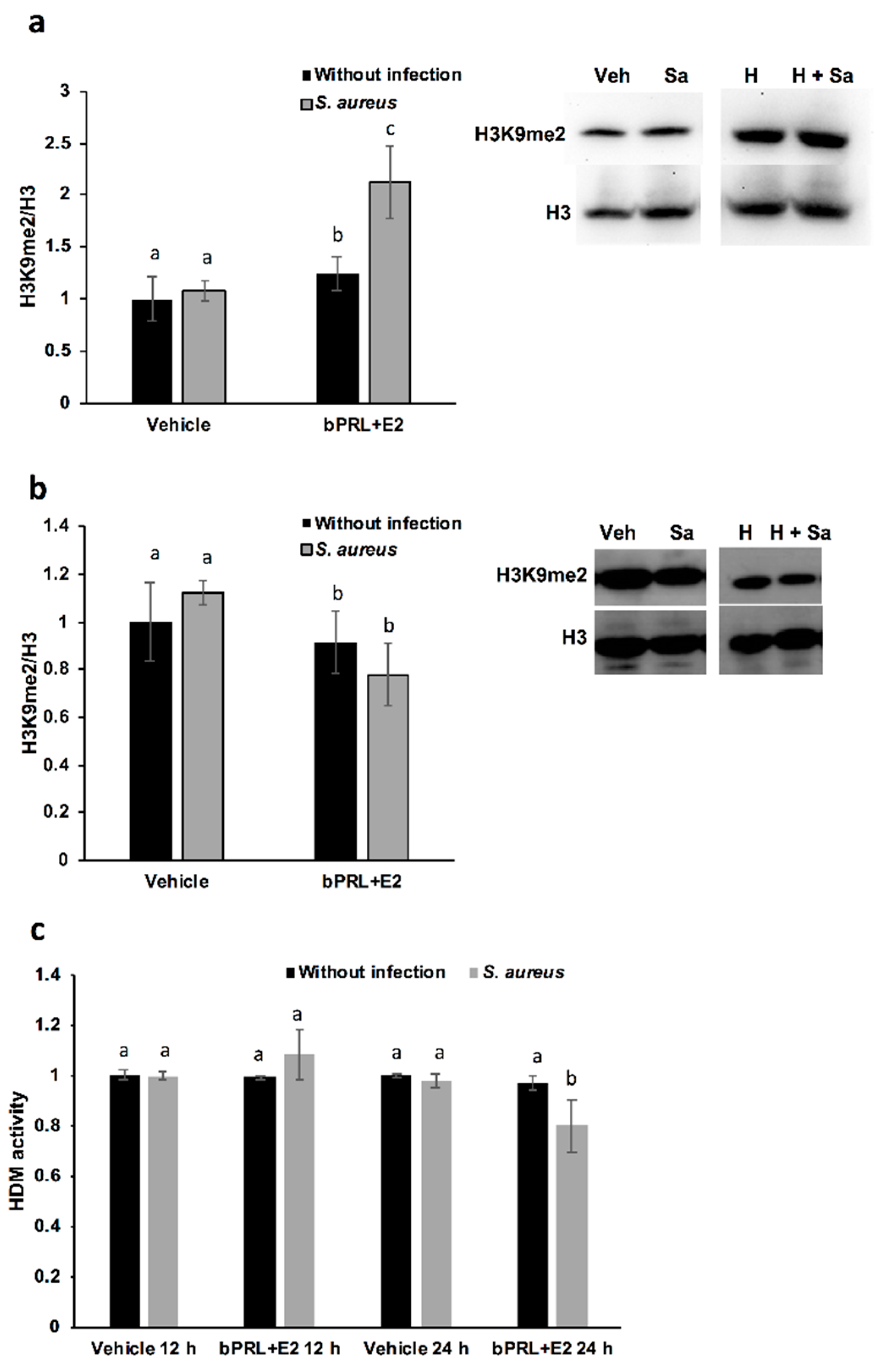
With respect to the epigenetic mark of H3K9me2, this is positively associated with hypoacetylation, therefore reducing DNA transcription [
25]. In this work, we demonstrated that this mark is induced in bMECs treated for 12 h with the combined hormones and infected, but at 24 h this mark is down-regulated under the same conditions. This is the first report that shows the role of this epigenetic mark in the bovine mammary epithelium. It is interesting that this mark coincides with H3K9Ac, as the combined hormones induce it in infected bMECs at 12 h, but at 24 h it is repressed. Altogether, these results suggest that the combined hormones can regulate the gene expression of inflammatory mediators through different epigenetic marks. In order to corroborate which genes are turned on or turned off by hormones, it is necessary to perform chromatin immunoprecipitation (ChIP) assays.
On the other hand, with the purpose of corroborating if HDMs are related to H3K9me2, we measure Jumonji family demethylases. Interestingly, HDM activity is reduced in bMECs treated for 24 h with the combined hormones and infected with
S. aureus, a result that coincides with the reduced H3K9me2 detected by a western blot under the same conditions. We attempt to correlate this activity with the levels of histone lysine demethylases (KDMs), in particular, with members of the family KDM4, which have high substrate preferences for lysine residues on histone H3. KDM4s can demethylate H3K9me2/3, H3K27me2/3 and H3K36me3 [
26]. In bovines, the role of these enzymes has been studied, mainly in fertilized oocytes [
27]. We detected a reduction in KDM4A gene expression at 12 h in bMECs treated with the combined hormones and infected, which could be responsible for reducing HDM activity under the same conditions. Finally, the epigenetic modulation of hormones in bMECs is interesting in terms of describing the novel effects of bPRL and E2. In this sense, the epigenetic modulation of PRL in bovine mammary glands has been mainly studied during mammary gland growth, development and differentiation, correlating epigenetic marks with milk protein production and differentiation [
28,
29]. Otherwise, studies related to the regulation of epigenetic marks in mammary glands by E2 have been mainly associated with the study of breast cancer [
30]. Thus, this work described, for the first time, that bPRL and E2 could be epigenetic modulators in bMECs and
S. aureus infection.
4. Materials and Methods
4.1. Reagents
Native purified bovine prolactin (bPRL) (lot AFP7170E) was provided by A. F. Parlow (NHPP, NIDDK, Torrance, CA, USA), dissolved in water and sterilized by filtration. Sigma-Aldrich (St. Louis, MO, USA) provided 17β-estradiol (E2), and working solutions were dissolved in 1% ethanol. For all of the experiments, 1% ethanol (vehicle) was used as a control. The hormone concentrations used in this work were bPRL 5 ng/mL and E2 50 pg/mL, as reported [
14,
15].
4.2. Antibodies
For flow cytometry analysis, the blocking antibody anti-α5β1 integrin (MAB2514) was purchased from Millipore (Burlington, MA, USA). The blocking anti-TLR2 (TL2.1) antibody was obtained from Abcam. Fluorescein isothiocyanate (FITC)-conjugated secondary antibodies against mouse or rat IgGs were purchased from Cell Signaling Technology.
For western blot analysis, the rabbit polyclonal antibody anti-H3Ac (Abcam, ab47915), the mouse monoclonal antibody anti-H3K9Ac (Santa Cruz, Sc56616), the mouse monoclonal antibody anti-H3K9me2 (Abcam, ab1220) and the rabbit polyclonal antibody anti-H3 (Abcam, ab1791) were used. Secondary antibodies raised against rabbit or mouse IgGs and coupled to horseradish peroxidase were obtained from Cell Signaling Technology (Danvers, MA, USA).
4.3. Staphylococcus aureus Strain
The
S. aureus subsp.
aureus (ATCC 27543) strain was used, which was isolated from a case of bovine clinical mastitis and has the capacity to invade bMECs [
31]. Bacteria were grown at 37 °C overnight in Luria–Bertani broth (LB, Bioxon), and the CFUs were adjusted by measuring the optical density at 600 nm (OD 0.2 = 9.2 × 10
7 CFU/mL).
4.4. Primary Culture of Bovine Mammary Epithelial Cells (bMECs)
bMECs were isolated from the alveolar tissue of the udders of healthy lactating cows (slaughtered for meat production), as described [
19]. Cells from passages 2–8 were used in all of the experiments. bMECs were cultured in growth medium (GM) composed by DMEM medium/nutrient mixture F12 Ham (DMEM/F12K, Sigma) and supplemented with 10% fetal calf serum (Equitech Bio), 10 μg/mL insulin (Sigma), 5 μg/mL hydrocortisone (Sigma), 100 U/mL penicillin, 100 μg/mL streptomycin and 1 μg/mL amphotericin B (Invitrogen, Carlsbad, CA, USA). bMECs were grown in a 5% CO
2 atmosphere at 37 °C. All of the experiments were achieved in DMEM/F12K without phenol red (Sigma).
4.5. bMEC Viability and S. aureus Growth Assays
To determine the effect of bPRL and E2 on bMEC viability, 1 × 104 cells were incubated with the combined hormones (5 ng/mL of bPRL together with 50 pg/mL of E2) in an incomplete medium for 24 h at 37 °C in 96-well plates. The bMEC viability was tested using the trypan blue exclusion assay and the cells were counted in an automated cell counter (BIO-RAD, TC20).
To analyze the effect of the combined hormones on S. aureus growth, 9.2 × 107 CFU/mL were cultured at 37 °C in LB broth and growth was evaluated turbidimetrically. To evaluate the S. aureus viability in the presence of the combined hormones, bacteria were grown (as previously described) in LB broth and were treated with the hormones for 2 or 24 h at 37 °C. Later, the bacteria were plated on LB agar in triplicate and were incubated overnight at 37 °C. The number of CFUs was determined by standard colony counting. Each experiment was performed three times in triplicate. The data represent the percentage of cell viability, considering the cells treated with vehicle as 100% (1% ethanol).
4.6. Invasion Assays
For the infection assays, bMECs were cultured in serum-free DMEM/F12K without phenol red (Sigma) and antibiotics (incomplete medium) for 24 h (~1 × 104 cells were cultured onto 96-well flat-bottomed dishes (Corning, Corning, NY, USA) treated with 6–10 μg/cm2 rat-tail type I collagen (Sigma)), and then bMECs were incubated with the hormones and/or infected. For the hormonal treatment, 5 ng/mL of bPRL, together with 50 pg/mL of E2 were added to incomplete medium for 24 h and then were infected with S. aureus (MOI 30:1 bacteria per cell). For this, the bMECs were inoculated with 9.2 × 107 CFU/mL and incubated for 2 h in 5% CO2 at 37 °C. Then, the cells were washed three times with PBS (pH 7.4) and incubated with incomplete medium that was supplemented with 80 μg/mL of gentamicin for 1 h at 37 °C to eliminate extracellular bacteria. Finally, the bMEC monolayers were detached with trypsin (0.05%)-EDTA (0.02%) (Sigma) and lysed with 250 μL of sterile distilled water. The bMEC lysates were diluted 100-fold, plated on LB agar in triplicate and incubated overnight at 37 °C. The number of CFUs was determined by the standard colony counting technique. The number of bMECs cultured in each well plate was calculated for each invasion assay using a hemocytometer. Each experiment was performed three times in triplicate. The data represent the normalized ratio of the CFU recovered per bMEC, considering the cells treated with vehicle as 1 (1% ethanol).
4.7. Analysis of Receptors by Flow Cytometry
The bMEC-polarized monolayers were cultured in 24-well dishes (Corning) and were treated with the combined hormones for 24 h and/or infected with S. aureus, as described above. After this, the cells were washed three times with PBS, detached with trypsin (0.05%)-EDTA (0.02%) (Sigma), centrifuged at 3200× g for 10 min at 4 °C and washed with PBS. The bMEC pellet was blocked with normal goat serum (5% in PBS, Pierce, Rockford, IL, USA) for 30 min at 4 °C with shaking, and then the cells were centrifuged and the pellet was incubated with the primary antibodies anti-TLR2 or anti-integrin, separately. To measure the TLR2 membrane abundance (MA), the cells were incubated with the antibody at a dilution of 1:50 (PBS containing BSA 0.1%) for 1 h at 4 °C with shaking. For the determination of integrin MA, the cells were incubated with the anti-α5β1 integrin at a concentration of 10 μg/mL for 2 h at 4 °C with shaking. In all of the cases, after the primary antibody incubation, the bMECs were washed three times with PBS and incubated with the respective secondary antibody (diluted 1:50) (FITC-conjugated anti-mouse IgGs for TLR2 or anti-rat IgGs for α5β1 integrin) for 1 h at 4 °C with shaking in the dark. The pellet was recovered by centrifugation, washed and fixed with paraformaldehyde (4%) for 10 min at 4 °C and finally washed three times with PBS. The cells were then suspended in 100 μL of PBS. The fluorescent signals of 10,000 events were measured and evaluated using the BD AccuriTM C6 cytometer, and analyzed with FlowJo software. Each experiment was performed three times in triplicate. The data represent the normalized receptor membrane abundance, considering the cells treated with vehicle as 1 (1% ethanol).
4.8. RNA Isolation and Gene Expression Analysis
To analyze the effects of the combined hormones and/or
S. aureus on the expression of the IIR genes of bMECs, monolayers of cells cultured in six-well plates with 6–10 μg/cm
2 rat-tail type I collagen (Sigma) were incubated with the hormones (24 h) and/or
S. aureus for 2 h (MOI 30:1). The same procedure was performed to evaluate the genes of the epigenetic enzymes, but for these approaches, bMECs were incubated with hormones for 12 or 24 h, and/or
S. aureus for 2 h. bMEC total RNA (5 µg) was extracted with the Trizol reagent (Invitrogen) according to the manufacturer’s instructions. Genomic DNA contamination was removed from RNA samples with DNase I treatment (Invitrogen). Then, cDNA was synthesized as described [
19]. The relative quantification of gene expression (qPCR) was performed using the comparative Ct method (∆∆Ct) in a StepOne Plus Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s instructions. The reactions were carried out with VeriQuest SYBR Green qPCR master mix (Affymetrix, Santa Clara, CA, USA). Specific primer pairs were acquired from Invitrogen and Elim Biopharm (Hayward, CA, USA) (
Table 4), and their specificity was determined by end point PCR. The GAPDH gene was used as an internal control. Each experiment was performed four times in duplicate and the ∆∆Ct value was obtained by setting the basal expression of vehicle-treated bMECs as RQ = 1.
4.9. Western Blot Analysis
To analyze epigenetic marks, the histone extraction of bMECs was performed according to the protocol described by Shechter et al. [
32] for the acid extraction of histones. Briefly, bMECs (3 × 10
6 cells) were incubated in Petri dishes at confluence, then the treatments were added (the combined hormones for 12 or 24 h and/or
S. aureus for 2 h). After this, the cells were washed three times with PBS, detached with trypsin (0.05%)-EDTA (0.02%) (Sigma), centrifuged at 3200×
g for 10 min at 4 °C and washed with PBS. The bMEC pellet was resuspended in 1 mL hypotonic lysis buffer and transferred to a 1.5-mL tube. Then, it was incubated for 30 min on a rotator at 4 °C to promote the hypotonic swelling of cells and lysis by mechanical shearing during rotation. The intact nuclei were pelleted by spinning in a cooled tabletop centrifuge at 15,400×
g, 10 min, 4 °C and were re-suspended in 400 μL 0.4 N H
2SO
4. To lysis nuclei, they were incubated on a rotator overnight and then were centrifuged to remove nuclear debris at 15,400×
g, 10 min. The supernatant was incubated with TCA (final concentration of 33%) to precipitate the histones; then, they were incubated overnight at 4 °C. Histones were pelleted by spinning in a cooled tabletop centrifuge at 15,400×
g, 10 min at 4 °C. Histone pellets were washed with ice-cold acetone, and centrifugated at 15,400×
g, 5 min at 4 °C. This step was repeated three times. Then, histones were air-dried for 20 min at room temperature. Histone pellets were dissolved in ddH
2O and stored frozen at −20 °C. Histones were resolved on a 15% SDS-PAGE gel to verify integrity and concentration before performing western blot analysis.
Proteins were transferred to PVDF membranes using a semi-dry transference system (Fisher Scientific); then were blocked with 5% non-fat milk in PBS for 4 h at 4 °C. After that, membranes were incubated with primary antibodies at 4 °C overnight: anti-H3Ac (1:1,000), anti-H3K9Ac (1:200), anti-H3K9me2 (1:1000) or the control antibody anti-H3 (1:5000). Membranes were incubated with anti-mouse or anti-rabbit horseradish peroxidase-coupled secondary antibodies (1:3000) for 2 h at 4 °C. Finally, ECL western blotting substrate WesternSureTM (Thermo Scientific, Waltham, MA, USA) was added and membranes were exposed to X-ray film or were developed using the iBright CL1500 Imaging System (Thermo Scientific). Signal intensity was quantified by densitometry using iBright Analysis Software (Thermo Scientific). Each western blot was performed at least three times from different experiments. The data represent the normalized ratio of the target histone mark with relation to H3 expression, considering the cells treated with vehicle as 1 (1% ethanol).
4.10. Histone Deacetylase (HDAC) Activity Assay
Plates of 96 wells were used to grow 1 × 105 bMECs with the combined hormones for 6 or 12 h, and then were infected with S. aureus for 2 h. HDAC activity was measured using a Fluor de Lys® kit (Enzo LifeSciences, Farmingdale, NY, USA) to analyze class I HDACs (HDAC1, 2, 3 and 8), IIa (HDAC4, 5, 7 and 9) and IIb (HDAC6 and 10). Then, bMECs were incubated with 2000 pmol of the acetylated Fluor de Lys® substrate for 4 h. Cells, media and the standard curve of the deacetylated substrate were prepared according to the manufacturer’s directions; relative fluorescence unit (RFU) counts were acquired with a Varioskan (Enzo LifeSciences) plate reader (360 nm excitation, 460 nm emission). A standard curve was generated using serial 1:10 dilutions of the deacetylated Fluor de Lys standard and developer supplied with the BML-AK503 HDAC fluorometric cellular activity assay kit. Trichostatin (TSA, 1 μM) was purchased from Sigma, and was used as an inhibitor of HDACs. HDAC activity was calculated, considering the effect of vehicle-treated bMECs as basal activity (normalized to onefold). According to the manufacturer’s instructions, the assays were run in duplicate from two different experiments.
4.11. Histone Demethylase (HDM) Activity Assay
We employed a kit from ThermoFisher (Cat. EIAHDMF) (Waltham, MA, USA) to measure the activity of Jumonji family demethylases (HDMs) in bMECs treated with the combined hormones and infected. Cells were grown in plates of 96 wells and treated as described for the HDAC assay, and cell lysates were obtained according to the manufacturer’s instructions. The product of the enzymatic demethylation reactions is formaldehyde, which was quantitated directly by a fluorescent product with a Varioskan (Thermo Scientific) plate reader (450 nm excitation, 510 nm emission). HDM activity was calculated, considering the effect of vehicle-treated bMECs as basal activity (normalized to onefold). According to the manufacturer’s instructions, the assays were run in duplicate from two different experiments.
4.12. Statistical Analysis
The data were analyzed in PRISM 8.0 software by performing a one-way analysis of variance (one-way ANOVA) and using the post hoc Tukey test. The results are reported as the means ± the standard errors (SE), and the significance level was set at
p ≤ 0.05, except for RT-qPCR analysis, where fold change values greater than two or less than 0.5 were considered as significantly differentially expressed mRNAs [
33].

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}