Genetic Diversity of Babesia bovis MSA-1, MSA-2b and MSA-2c in China

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
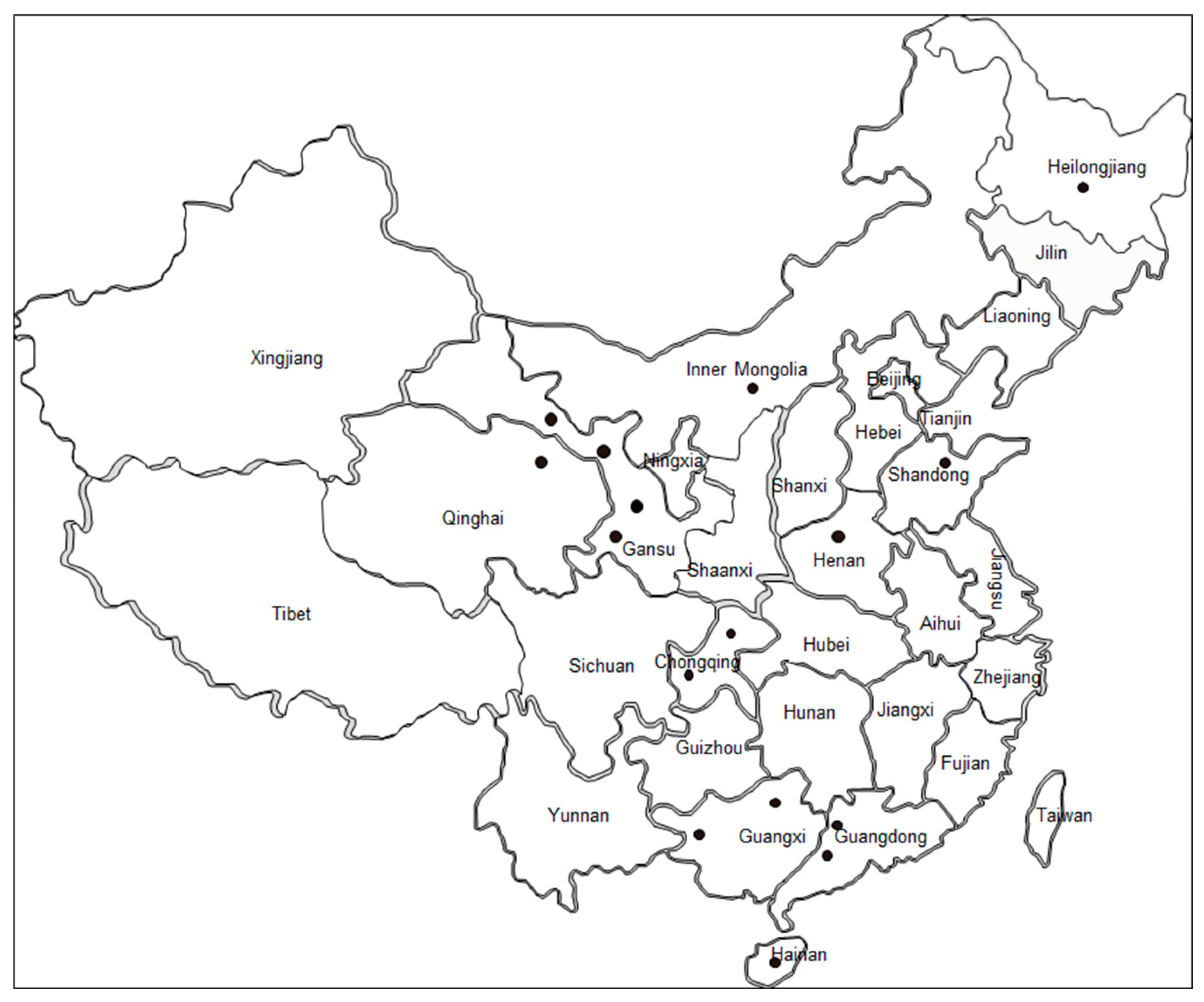
2.1. Sample Collection and DNA Extraction
2.2. Screening Positive Samples for B. bovis Infection
2.3. MSA-1, MSA-2b and MSA-2b Gene Amplification and Sequencing
2.4. Phylogenetic Analyses
3. Results
3.1. Diagnosis of B. bovis Infection in Cattle
3.2. DNA Sequencing of MSA-1, MSA-2b, and MSA-2c Genes
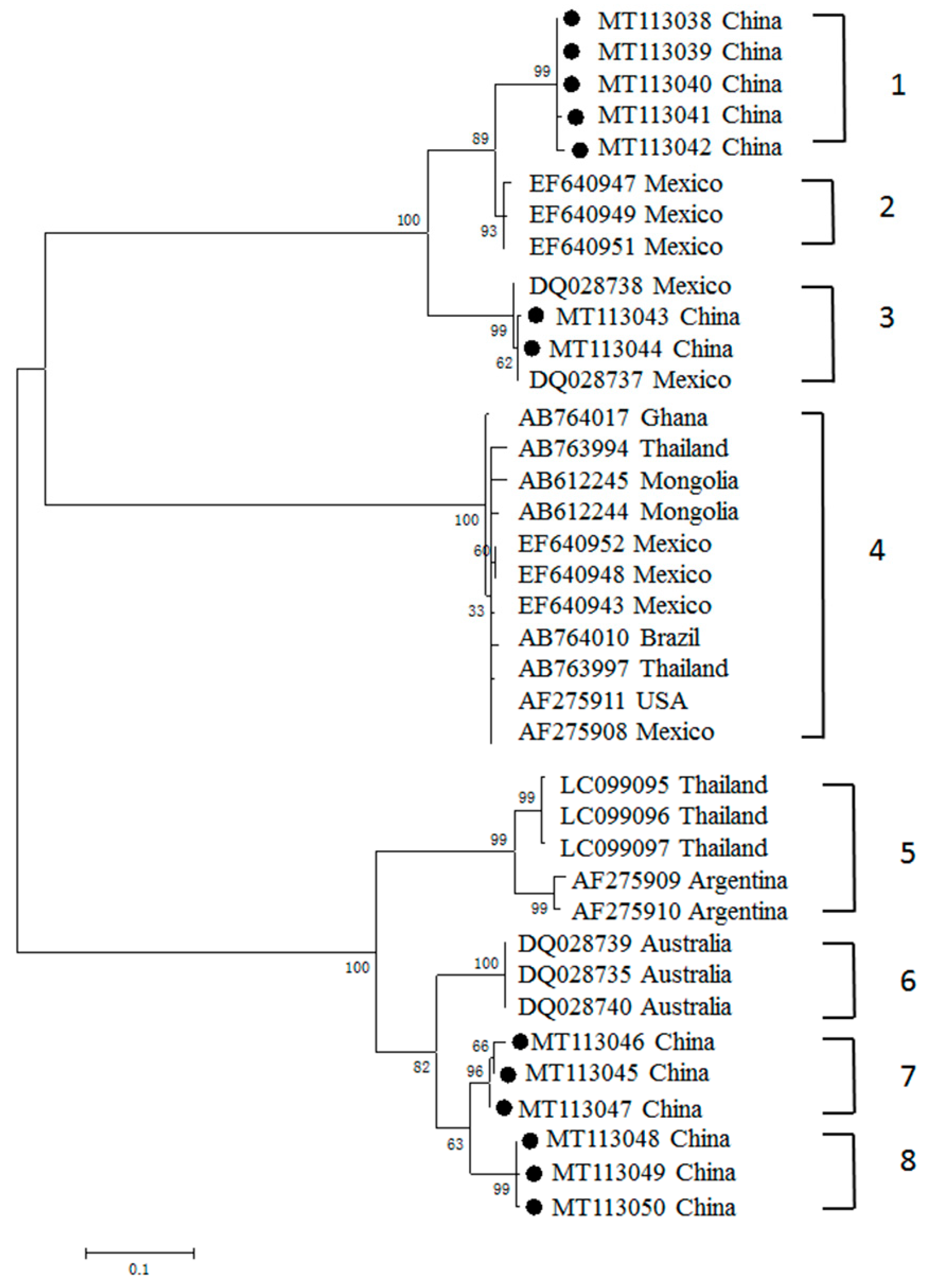
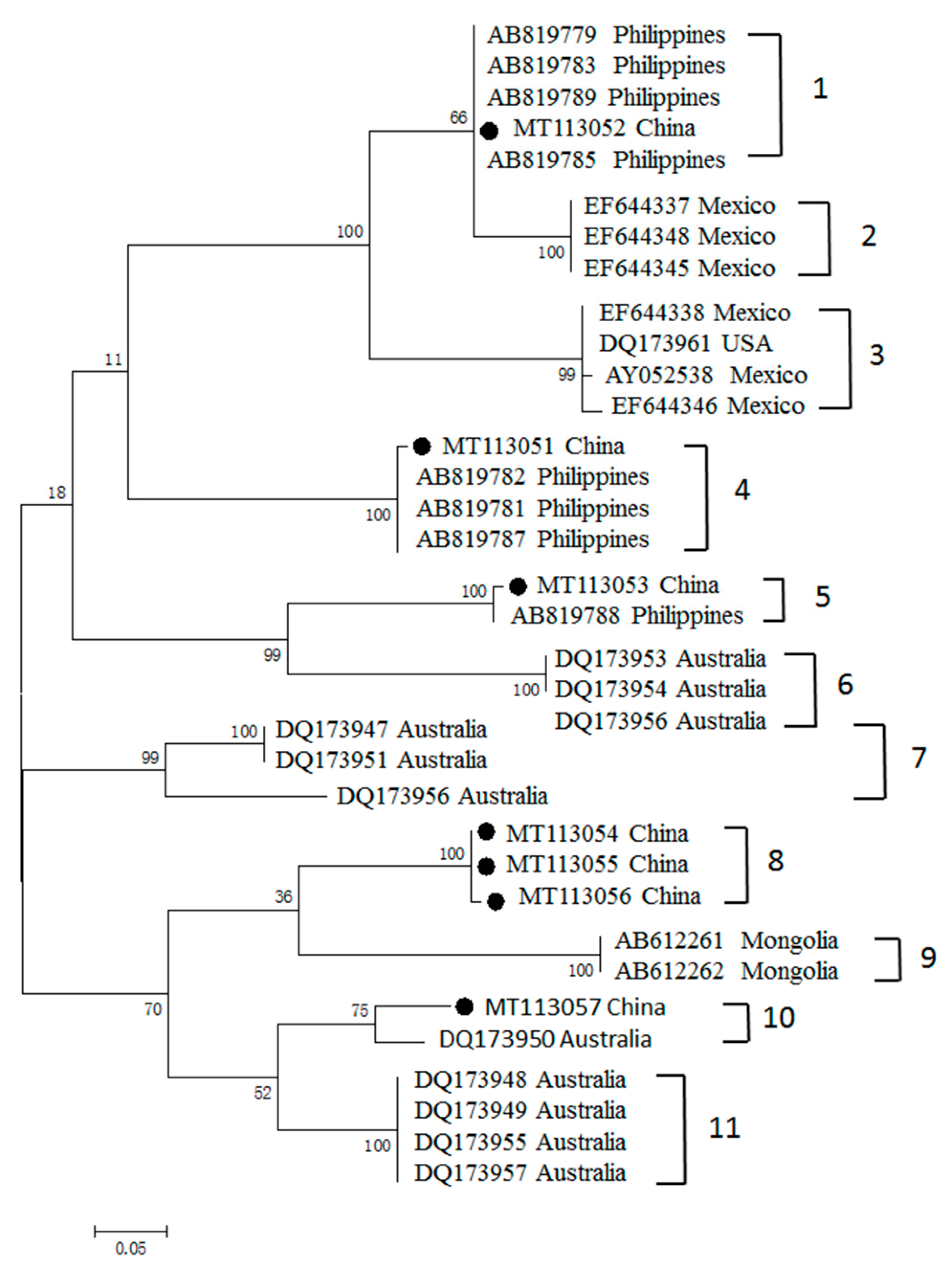
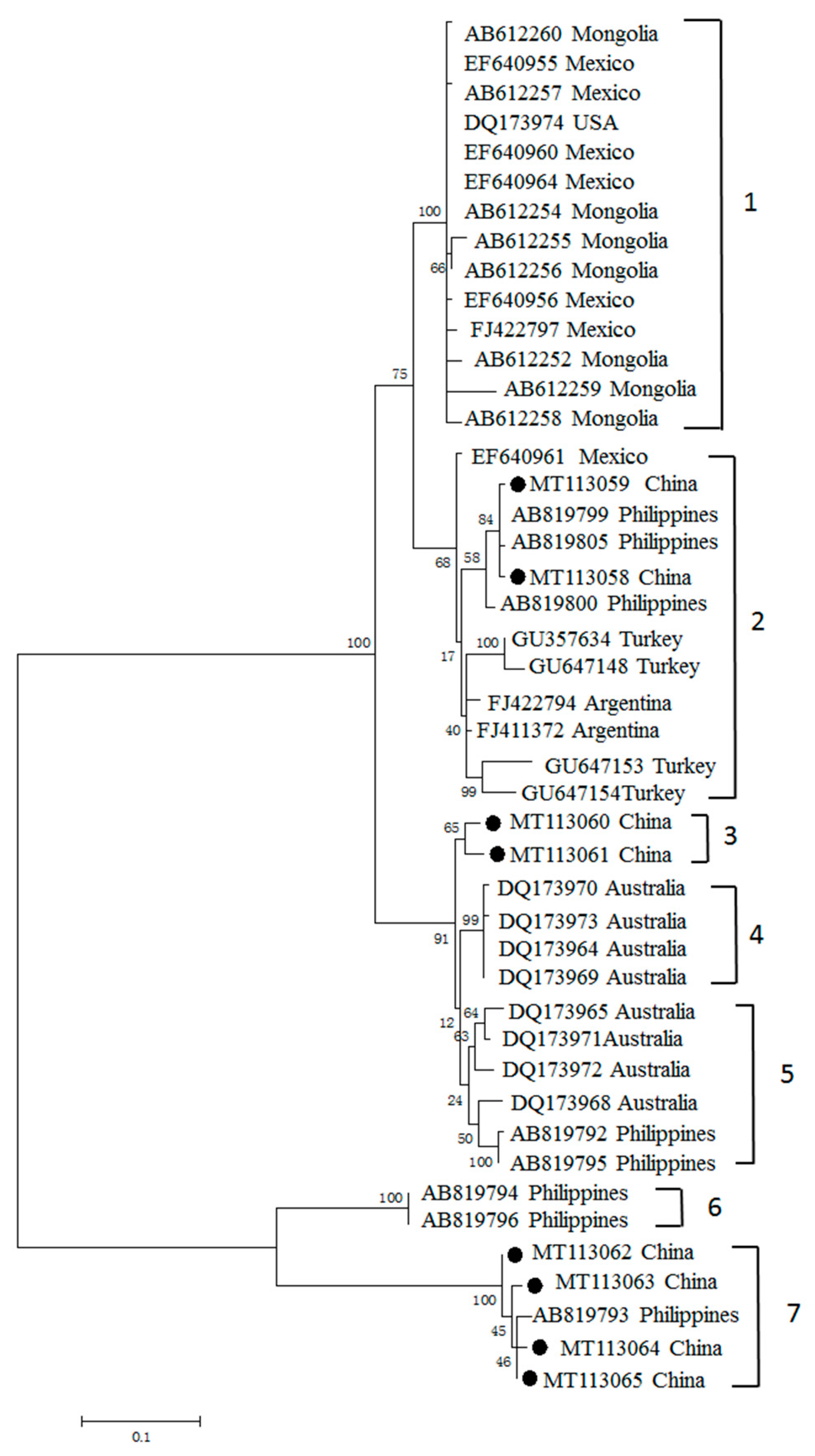
3.3. Phylogenetic Analyses of MSA-1, MSA-2b and MSA-2c Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bock, R.; Jackson, L.; de Vos, A.; Jorgensen, W. Babesiosis of cattle. Parasitology 2004, 129, S247–S269. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.C.; Norimine, J.; Goff, W.L.; Suarez, C.E.; McElwain, T.F. Prospects for recombinant vaccines against Babesia bovis and related parasites. Parasite Immunol. 2006, 28, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Lu, W.; Luo, J. Babesiosis in China. Trop. Anim. Health Prod. 1997, 29, 11S–15S. [Google Scholar] [CrossRef] [PubMed]
- Niu, Q.; Liu, Z.; Yu, P.; Yang, J.; Abdallah, M.O.; Guan, G.; Liu, G.; Luo, J.; Yin, H. Genetic characterization and molecular survey of Babesia bovis, Babesia bigemina and Babesia ovata in cattle, dairy cattle and yaks in China. Parasites Vectors 2015, 8, 518. [Google Scholar] [CrossRef]
- Liu, J.; Guan, G.; Liu, A.; Li, Y.; Yin, H.; Luo, J. A PCR method targeting internal transcribed spacers: The simultaneous detection of Babesia bigemina and Babesia bovis in cattle. Acta Parasitol. 2014, 59, 132–138. [Google Scholar] [CrossRef]
- Luo, J.; Yin, H.; Guan, G.; Zhang, Q.; Lu, W. Description of a new Babesia sp. infective for cattle in China. Parasitol. Res. 2002, 88, S13–S15. [Google Scholar] [CrossRef]
- Berens, S.J.; Brayton, K.A.; Molloy, J.B.; Bock, R.E.; Lew, A.E.; McElwain, T.F. Merozoite surface antigen 2 proteins of Babesia bovis vaccine breakthrough isolates contain a unique hypervariable region composed of degenerate repeats. Infect. Immun. 2005, 73, 7180–7189. [Google Scholar] [CrossRef]
- Yokoyama, N.; Okamura, M.; Igarashi, I. Erythrocyte invasion by Babesia parasites: Current advances in the elucidation of the molecular interactions between the protozoan ligands and host receptors in the invasion stage. Vet. Parasitol. 2006, 138, 22–32. [Google Scholar] [CrossRef]
- Hines, S.A.; McElwain, T.F.; Buening, G.M.; Palmer, G.H. Molecular characterization of Babesia bovis merozoite surface proteins bearing epitopes immunodominant in protected cattle. Mol. Biochem. Parasitol. 1989, 37, 1–9. [Google Scholar] [CrossRef]
- Jasmer, D.P.; Reduker, D.W.; Hines, S.A.; Perryman, L.E.; McGuire, T.C. Surface epitope localization and gene structure of a Babesia bovis 44-kilodalton variable merozoite surface antigen. Mol. Biochem. Parasitol. 1992, 55, 75–83. [Google Scholar] [CrossRef]
- Mosqueda, J.; McElwain, T.F.; Palmer, G.H. Babesia bovis merozoite surface antigen 2 proteins are expressed on the merozoite and sporozoite surface, and specific antibodies inhibit attachment and invasion of erythrocytes. Infect. Immun. 2002, 70, 6448–6455. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Suarez, C.E.; Florin-Christensen, M.; Hines, S.A.; Palmer, G.H.; Brown, W.C.; McElwain, T.F. Characterization of allelic variation in the Babesia bovis merozoite surface antigen 1 (MSA-1) locus and identification of a cross-reactive inhibition-sensitive MSA-1 epitope. Infect. Immun. 2000, 68, 6865–6870. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Florin-Christensen, M.; Suarez, C.E.; Hines, S.A.; Palmer, G.H.; Brown, W.C.; McElwain, T.F. The Babesia bovis merozoite surface antigen 2 locus contains four tandemly arranged and expressed genes encoding immunologically distinct proteins. Infect. Immun. 2002, 70, 3566–3575. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hines, S.A.; Palmer, G.H.; Jasmer, D.P.; McGuire, T.C.; McElwain, T.F. Neutralization-sensitive merozoite surface antigens of Babesia bovis encoded by members of a polymorphic gene family. Mol. Biochem. Parasitol. 1992, 55, 85–94. [Google Scholar] [CrossRef]
- Hines, S.A.; Palmer, G.H.; Jasmer, D.P.; Goff, W.L.; McElwain, T.F. Immunization of cattle with recombinant Babesia bovis merozoite surface antigen-1. Infect. Immun. 1995, 63, 349–352. [Google Scholar] [CrossRef] [PubMed]
- McElwain, T.F.; Hines, S.A.; Palmer, G.H. Persistence of antibodies against epitopes encoded by a single gene copy of the Babesia bovis merozoite surface antigen 1 (MSA-1). J. Parasitol. 1998, 84, 449–452. [Google Scholar] [CrossRef]
- Allred, D.R.; Hines, S.A.; Ahrens, K.P. Isolate-specific parasite antigens of the Babesia bovis-infected erythrocyte surface. Mol. Biochem. Parasitol. 1993, 60, 121–132. [Google Scholar] [CrossRef]
- Lau, A.O.T.; Cereceres, K.; Palmer, G.H.; Fretwell, D.L.; Pedroni, M.J.; Mosqueda, J.; Mcelwain, T.F. Genotypic diversity of merozoite surface antigen 1 of Babesia bovis within an endemic population. Mol. Biochem. Parasitol. 2010, 172, 107–112. [Google Scholar] [CrossRef]
- Nagano, D.; Sivakumar, T.; de de Macedo, A.C.C.; Inpankaew, T.; Alhassan, A.; Igarashi, I.; Yokoyama, N. The genetic diversity of merozoite surface antigen 1 (MSA-1) among Babesia bovis detected from cattle populations in Thailand, Brazil and Ghana. J. Vet. Med. Sci. 2013, 75, 1463–1470. [Google Scholar] [CrossRef]
- Tattiyapong, M.; Sivakumar, T.; Ybanez, A.P.; Ybanez, R.H.D.; Perez, Z.O.; Guswanto, A.; Igarashi, I.; Yokoyama, N. Diversity of Babesia bovis merozoite surface antigen genes in the Philippines. Parasitol. Int. 2014, 63, 57–63. [Google Scholar] [CrossRef]
- Tattiyapong, M.; Sivakumar, T.; Takemae, H.; Simking, P.; Jittapalapong, S.; Igarashi, I.; Yokoyama, N. Genetic diversity and antigenicity variation of Babesia bovis merozoite surface antigen-1 (MSA-1) in Thailand. Infect. Genet. Evol. 2016, 41, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Matos, C.A.; Goncalves, L.R.; Alvarez, D.O.; Freschi, C.R.; da Silva, J.B.; Valmoraes, S.P.; Mendes, N.S.; Andre, M.R.; Machado, R.Z. Longitudinal evaluation of humoral immune response and merozoite surface antigen diversity in calves naturally infected with Babesia bovis, in Sao Paulo, Brazil. Rev. Bras. Parasitol. Vet. 2017, 26, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Altangerel, K.; Sivakumar, T.; Battsetseg, B.; Battur, B.; Ueno, A.; Igarashi, I.; Yokoyama, N. Phylogenetic relationships of Mongolian Babesia bovis isolates based on the merozoite surface antigen (MSA)-1, MSA-2b, and MSA-2c genes. Vet. Parasitol. 2012, 184, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, M.; Echaide, I.; de Echaide, S.T.; Mosqueda, J.; Cetra, B.; Suarez, C.E.; Florinchristensen, M. In silico predicted conserved B-cell epitopes in the merozoite surface antigen-2 family of B. bovis are neutralization sensitive. Vet. Parasitol. 2010, 167, 216–226. [Google Scholar] [CrossRef]
- Leroith, T.; Brayton, K.A.; Molloy, J.B.; Bock, R.E.; Hines, S.A.; Lew, A.E.; Mcelwain, T.F. Sequence variation and immunologic cross-reactivity among Babesia bovis merozoite surface antigen 1 proteins from vaccine strains and vaccine breakthrough isolates. Infect. Immun. 2005, 73, 5388–5394. [Google Scholar] [CrossRef]
- Borgonio, V.M.; Mosqueda, J.; Genis, A.D.; Falcon, A.; Alvarez, J.A.; Camacho, M.; Figueroa, J.V. msa-1 and msa-2c Gene Analysis and Common Epitopes Assessment in Mexican Babesia bovis Isolates. Ann. N. Y. Acad. Sci. 2008, 1149, 145–148. [Google Scholar] [CrossRef]
- AbouLaila, M.; Yokoyama, N.; Igarashi, I. Development and evaluation of a nested PCR based on spherical body protein 2 gene for the diagnosis of Babesia bovis infection. Vet. Parasitol. 2010, 169, 45–50. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Li, Y.; Li, J.; Chahan, B.; Guo, Q.; Zhang, Y.; Moumouni, P.F.A.; Lee, S.; Liu, M.; Galon, E.M.; Guo, H.; et al. Molecular investigation of tick-borne infections in cattle from Xinjiang Uygur Autonomous Region, China. Parasitol. Int. 2020, 74, 101925. [Google Scholar] [CrossRef]
- Mendes, N.S.; Ramos, I.A.D.; Herrera, H.M.; Campos, J.B.V.; Alves, J.V.D.; de Macedo, G.C.; Machado, R.Z.; Andre, M.R. Genetic diversity of Babesia bovis in beef cattle in a large wetland in Brazil. Parasitol. Res. 2019, 118, 2027–2040. [Google Scholar] [CrossRef]
- Liyanagunawardena, N.; Sivakumar, T.; Kothalawala, H.; Silva, S.S.P.; Battsetseg, B.; Lan, D.T.B.; Inoue, N.; Igarashi, I.; Yokoyama, N. Type-specific PCR assays for Babesia bovis msa-1 genotypes in Asia: Revisiting the genetic diversity in Sri Lanka, Mongolia, and Vietnam. Infect. Genet. Evol. 2016, 37, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Simuunza, M.; Bilgic, H.B.; Karagenc, T.; Syakalima, M.; Shiels, B.; Tait, A.; Weir, W. Population genetic analysis and sub-structuring in Babesia bovis. Mol. Biochem. Parasitol. 2011, 177, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Oura, C.A.L.; Asiimwe, B.B.; Weir, W.; Lubega, G.W.; Tait, A. Population genetic analysis and sub-structuring of Theileria parva in Uganda. Mol. Biochem. Parasitol. 2005, 140, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Weir, W.; Karagenc, T.; Gharbi, M.; Simuunza, M.; Aypak, S.; Aysul, N.; Darghouth, M.A.; Shiels, B.; Tait, A. Population diversity and multiplicity of infection in Theileria annulata. Int. J. Parasitol. 2011, 41, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, J.; Liu, J.; Wang, X.; Xu, J.; Liu, A.; Li, Y.; Liu, Z.; Ren, Q.; Luo, J.; et al. Molecular detection and genetic diversity of Theileria orientalis in cattle in China. Parasitol. Res. 2018, 117, 3689–3694. [Google Scholar] [CrossRef]
- Eamens, G.J.; Gonsalves, J.R.; Jenkins, C.; Collins, D.; Bailey, G. Theileria orientalis MPSP types in Australian cattle herds associated with outbreaks of clinical disease and their association with clinical pathology findings. Vet. Parasitol. 2013, 191, 209–217. [Google Scholar] [CrossRef]
- Kim, S.; Yu, D.; Chae, J.; Choi, K.; Kim, H.; Park, B.; Chae, J.; Park, J. Pathogenic genotype of major piroplasm surface protein associated with anemia in Theileria orientalis infection in cattle. Acta Vet. Scand. 2017, 59, 51. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Province | No. of Samples | No. of Positive Samples (%) |
|---|---|---|
| Hainan | 59 | 6 (10.2%) |
| Qinghai | 39 | 3 (7.7%) |
| Guangxi | 71 | 1 (1.4%) |
| Guangdong | 59 | 9 (15.3%) |
| Gansu | 94 | 5 (5.3%) |
| Heilongjiang | 33 | 3 (9.1%) |
| Chongqing | 51 | 5 (9.8%) |
| Shandong | 49 | 2 (4.1) |
| Henan | 56 | 1 (1.8%) |
| Inner Mongolia | 64 | 16 (25%) |
| Total | 575 | 51 (8.9%) |
| Province | Sample ID | Gene Accession No. | ||
|---|---|---|---|---|
| MSA-1 | MSA-2b | MSA-2c | ||
| Hainan | Han2 | MT113038 | MT113051 | |
| Han13 | MT113052 | MT113058 | ||
| Han4 | MT113039 | MT113059 | ||
| Qinghai | QH19 | MT113053 | ||
| Guangxi | GXBS3 | MT113040 | MT113054 | |
| Guangdong | GDMM23 | MT113041 | MT113060 | |
| GDMM44 | MT113042, MT113050 | |||
| GDMM53 | MT113061 | |||
| GDMM59 | MT113043 | |||
| Gansu | GSYZ17 | MT113044 | MT113062 | |
| GSQL1326 | MT113055 | |||
| GSQL1330 | MT113063 | |||
| Heilongjiang | HLJ795 | MT113045 | ||
| HLJL22 | MT113046 | |||
| Chongqing | CQBY32 | MT113056 | ||
| CQJJ11 | MT113064 | |||
| Shandong | SDJN7 | MT113047 | ||
| SDJN16 | MT113057 | |||
| Inner Mongolia | IM27 | MT113048 | ||
| IM39 | MT113049 | |||
| IM7 | MT113050 | MT113065 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Yang, J.; Gao, S.; Wang, X.; Sun, H.; Lv, Z.; Li, Y.; Liu, A.; Liu, J.; Luo, J.; et al. Genetic Diversity of Babesia bovis MSA-1, MSA-2b and MSA-2c in China. Pathogens 2020, 9, 473. https://doi.org/10.3390/pathogens9060473
Wang J, Yang J, Gao S, Wang X, Sun H, Lv Z, Li Y, Liu A, Liu J, Luo J, et al. Genetic Diversity of Babesia bovis MSA-1, MSA-2b and MSA-2c in China. Pathogens. 2020; 9(6):473. https://doi.org/10.3390/pathogens9060473
Chicago/Turabian StyleWang, Jinming, Jifei Yang, Shandian Gao, Xiaoxing Wang, Hao Sun, Zhaoyong Lv, Youquan Li, Aihong Liu, Junlong Liu, Jianxun Luo, and et al. 2020. "Genetic Diversity of Babesia bovis MSA-1, MSA-2b and MSA-2c in China" Pathogens 9, no. 6: 473. https://doi.org/10.3390/pathogens9060473
APA StyleWang, J., Yang, J., Gao, S., Wang, X., Sun, H., Lv, Z., Li, Y., Liu, A., Liu, J., Luo, J., Guan, G., & Yin, H. (2020). Genetic Diversity of Babesia bovis MSA-1, MSA-2b and MSA-2c in China. Pathogens, 9(6), 473. https://doi.org/10.3390/pathogens9060473