Description of Virulent Factors and Horizontal Gene Transfers of Keratitis-Associated Amoeba Acanthamoeba Triangularis by Genome Analysis


Abstract
1. Introduction
2. Results
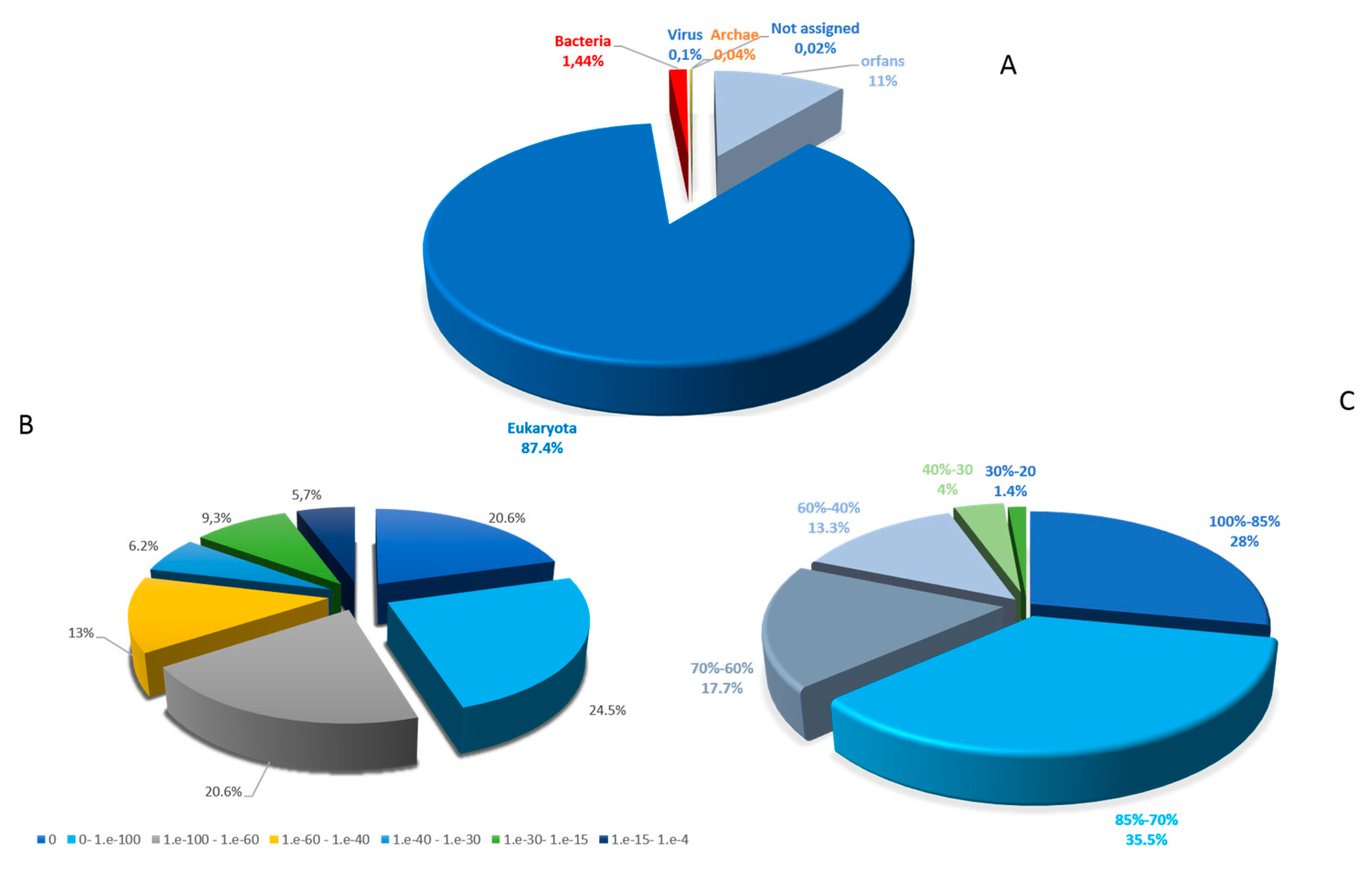
2.1. General Genomic Features
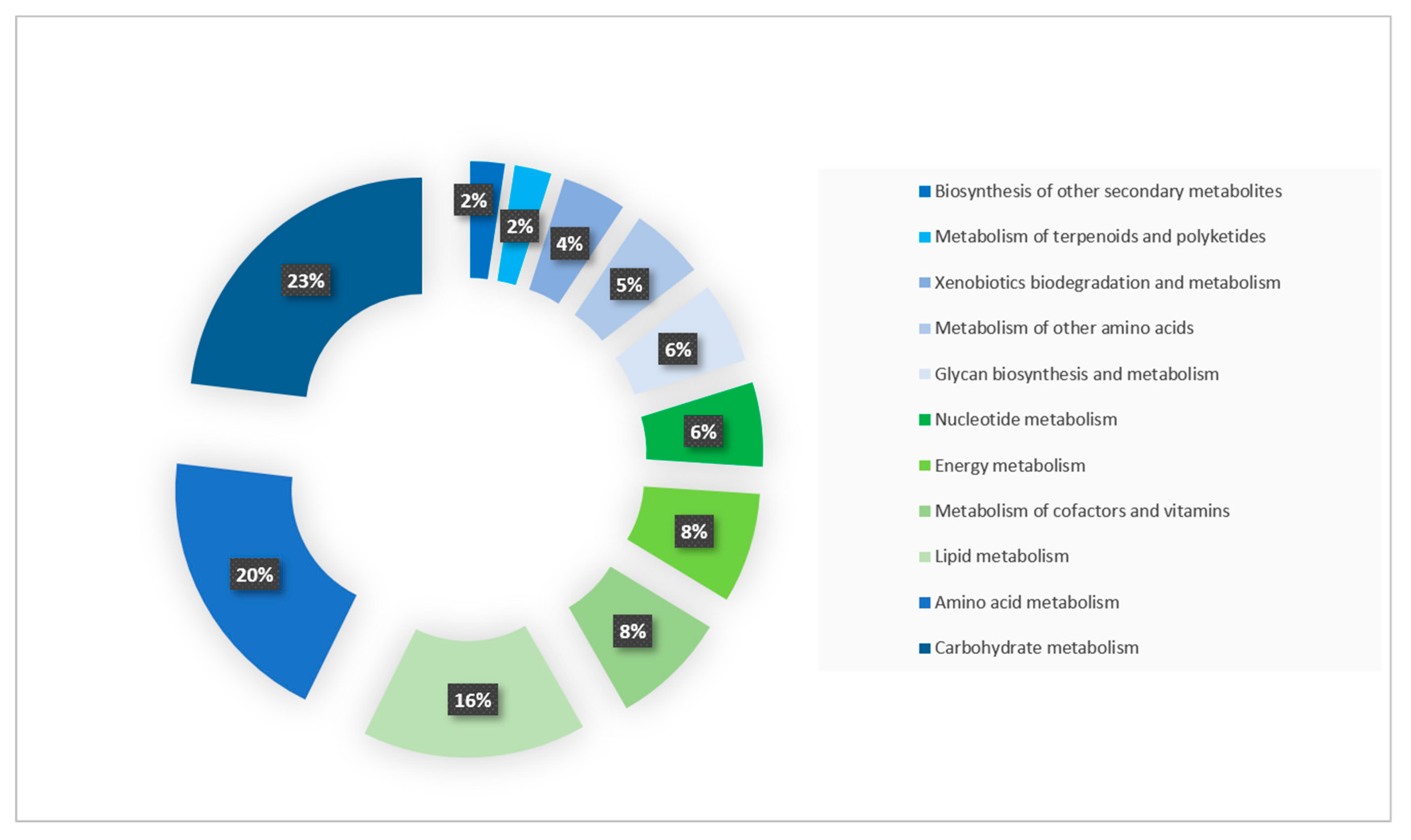
2.2. Functional Annotation
2.3. Gene Related to Keratitis Pathogenicity
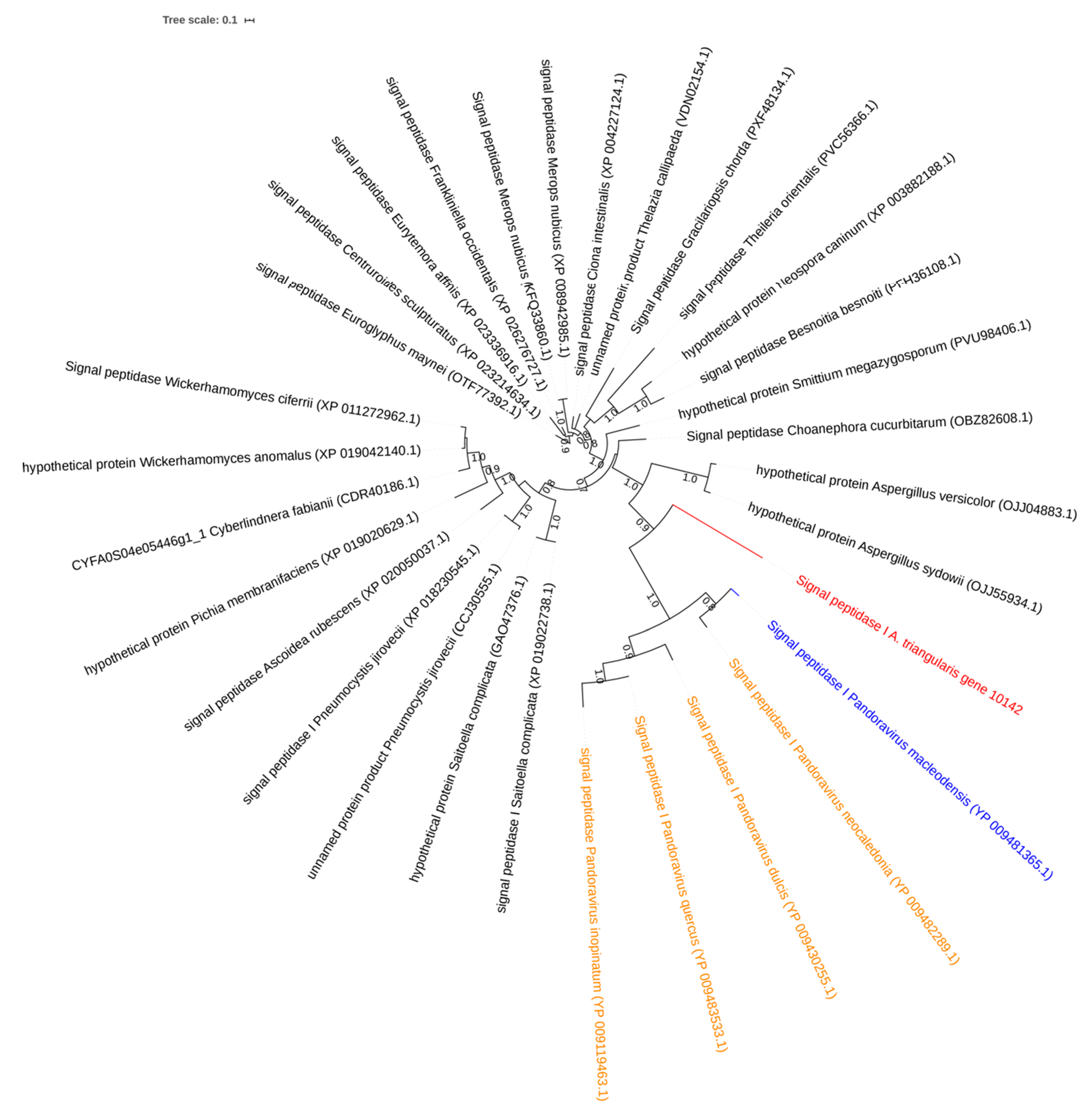
2.4. Investigation on Sequences Inherited from Potential Horizontal Gene Gransfers
2.5. Pan-Genome and Core Genome Analyses of Acanthamoeba spp.
3. Discussion
4. Materials and Methods
4.1. Culture of Acanthamoeba Triangularis Strain SH 621
4.2. Extraction and Sequencing of DNA
4.3. Genome Assembly
4.4. Functional Annotation
4.5. Analysis of Virulence Related Genes
4.6. Study of Horizontal Gene Transfers Between A. triangularis and ARMs
4.7. Comparative Genomic Analyses
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rodríguez-Zaragoza, S. Ecology of free-living amoebae. Crit. Rev. Microbiol. 1994, 20, 225–241. [Google Scholar] [CrossRef]
- Khan, N.A. Acanthamoeba: Biology and increasing importance in human health. FEMS Microbiol. Rev. 2006, 30, 564–595. [Google Scholar] [CrossRef]
- Siddiqui, R.; Khan, N.A. Biology and pathogenesis of Acanthamoeba. Parasites Vectors 2012, 5, 6. [Google Scholar] [CrossRef]
- Clarke, D.W.; Niederkorn, J.Y. The pathophysiology of Acanthamoeba keratitis. Trends Parasitol. 2006, 22, 175–180. [Google Scholar] [CrossRef]
- Kot, K.; Łanocha-Arendarczyk, N.A.; Kosik-Bogacka, D.I. Amoebas from the genus Acanthamoeba and their pathogenic properties. Ann. Parasitol. 2018, 64, 299–308. [Google Scholar]
- Lorenzo-Morales, J.; Khan, N.A.; Walochnik, J. An update on Acanthamoeba keratitis: Diagnosis, pathogenesis and treatment. Parasite 2015, 22. [Google Scholar] [CrossRef]
- Trabelsi, H.; Dendana, F.; Sellami, A.; Sellami, H.; Cheikhrouhou, F.; Neji, S.; Makni, F.; Ayadi, A. Pathogenic free-living amoebae: Epidemiology and clinical review. Pathol. Biol. 2012, 60, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Taher, E.E.; Méabed, E.M.H.; Abdallah, I.; Abdel Wahed, W.Y. Acanthamoeba keratitis in noncompliant soft contact lenses users: Genotyping and risk factors, a study from Cairo, Egypt. J. Infect. Public Health 2018, 11, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Castro-Artavia, E.; Retana-Moreira, L.; Lorenzo-Morales, J.; Abrahams-Sandí, E. Potentially pathogenic Acanthamoeba genotype T4 isolated from dental units and emergency combination showers. Mem. Inst. Oswaldo Cruz 2017, 112, 817–821. [Google Scholar] [CrossRef] [PubMed]
- Xuan, Y.-H.; Chung, B.-S.; Hong, Y.-C.; Kong, H.-H.; Hahn, T.-W.; Chung, D.-I. Keratitis by acanthamoeba triangularis: Report of cases and characterization of isolates. Korean J. Parasitol. 2008, 46, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Anwar, A.; Khan, N.A.; Siddiqui, R. Combating Acanthamoeba spp. cysts: What are the options? Parasit Vectors 2018, 11, 26. [Google Scholar] [CrossRef] [PubMed]
- Clarholm, M. Protozoan grazing of bacteria in soil—Impact and importance. Microb. Ecol. 1981, 7, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Greub, G.; Raoult, D. Microorganisms resistant to free-living amoebae. Clin. Microbiol. Rev. 2004, 17, 413–433. [Google Scholar] [CrossRef] [PubMed]
- Beye, M.; Hasni, I.; Seng, P.; Michelle, C.; La Scola, B.; Raoult, D.; Fournier, P.-E. Genomic analysis of a Raoultella ornithinolytica strain causing prosthetic joint infection in an immunocompetent patient. Sci. Rep. 2018, 8, 9462. [Google Scholar] [CrossRef]
- Hasni, I.; Jarry, A.; Quelard, B.; Carlino, A.; Eberst, J.-B.; Abbe, O.; Demanèche, S. Intracellular behaviour of three legionella pneumophila strains within three amoeba strains, including willaertia magna C2c maky. Pathogens 2020, 9, 105. [Google Scholar] [CrossRef]
- Andreani, J.; Khalil, J.Y.B.; Baptiste, E.; Hasni, I.; Michelle, C.; Raoult, D.; Levasseur, A.; La Scola, B. Orpheovirus IHUMI-LCC2: A New Virus among the Giant Viruses. Front. Microbiol. 2018, 8, 2643. [Google Scholar] [CrossRef]
- Moliner, C.; Raoult, D.; Fournier, P.-E. Evidence of horizontal gene transfer between amoeba and bacteria. Clin. Microbiol. Infect. 2009, 15, 178–180. [Google Scholar] [CrossRef]
- Bertelli, C.; Greub, G. Lateral gene exchanges shape the genomes of amoeba-resisting microorganisms. Front. Cell Infect. Microbiol. 2012, 2, 110. [Google Scholar] [CrossRef]
- Clarke, M.; Lohan, A.J.; Liu, B.; Lagkouvardos, I.; Roy, S.; Zafar, N.; Bertelli, C.; Schilde, C.; Kianianmomeni, A.; Bürglin, T.R.; et al. Genome of Acanthamoeba castellanii highlights extensive lateral gene transfer and early evolution of tyrosine kinase signaling. Genome Biol. 2013, 14, R11. [Google Scholar] [CrossRef]
- Karlyshev, A.V. Remarkable features of mitochondrial dna of acanthamoeba polyphaga linc ap-1, revealed by whole-genome sequencing. Microbiol. Resour. Announc. 2019, 8, e00430-19. [Google Scholar] [CrossRef]
- Chelkha, N.; Levasseur, A.; Pontarotti, P.; Raoult, D.; Scola, B.L.; Colson, P. A phylogenomic study of acanthamoeba polyphaga draft genome sequences suggests genetic exchanges with giant viruses. Front. Microbiol. 2018, 9, 2098. [Google Scholar] [CrossRef] [PubMed]
- Zysset-Burri, D.C.; Müller, N.; Beuret, C.; Heller, M.; Schürch, N.; Gottstein, B.; Wittwer, M. Genome-wide identification of pathogenicity factors of the free-living amoeba Naegleria fowleri. BMC Genom. 2014, 15, 496. [Google Scholar] [CrossRef] [PubMed]
- Fritz-Laylin, L.K.; Prochnik, S.E.; Ginger, M.L.; Dacks, J.B.; Carpenter, M.L.; Field, M.C.; Kuo, A.; Paredez, A.; Chapman, J.; Pham, J.; et al. The genome of naegleria gruberi illuminates early eukaryotic versatility. Cell 2010, 140, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Liechti, N.; Schürch, N.; Bruggmann, R.; Wittwer, M. The genome of Naegleria lovaniensis, the basis for a comparative approach to unravel pathogenicity factors of the human pathogenic amoeba N. fowleri. BMC Genom. 2018, 19, 654. [Google Scholar] [CrossRef]
- Sucgang, R.; Kuo, A.; Tian, X.; Salerno, W.; Parikh, A.; Feasley, C.L.; Dalin, E.; Tu, H.; Huang, E.; Barry, K.; et al. Comparative genomics of the social amoebae Dictyostelium discoideum and Dictyostelium purpureum. Genome Biol. 2011, 12, R20. [Google Scholar] [CrossRef]
- Lorenzi, H.A.; Puiu, D.; Miller, J.R.; Brinkac, L.M.; Amedeo, P.; Hall, N.; Caler, E.V. New assembly, reannotation and analysis of the entamoeba histolytica genome reveal new genomic features and protein content information. PLoS Negl. Trop. Dis. 2010, 4, e716. [Google Scholar] [CrossRef]
- Huth, S.; Reverey, J.F.; Leippe, M.; Selhuber-Unkel, C. Adhesion forces and mechanics in mannose-mediated acanthamoeba interactions. PLoS ONE 2017, 12, e0176207. [Google Scholar] [CrossRef]
- Guimaraes, A.J.; Gomes, K.X.; Cortines, J.R.; Peralta, J.M.; Peralta, R.H.S. Acanthamoeba spp. as a universal host for pathogenic microorganisms: One bridge from environment to host virulence. Microbiol. Res. 2016, 193, 30–38. [Google Scholar] [CrossRef]
- Yoshikawa, G.; Blanc-Mathieu, R.; Song, C.; Kayama, Y.; Mochizuki, T.; Murata, K.; Ogata, H.; Takemura, M. Medusavirus, a novel large DNA virus discovered from hot spring water. J. Virol. 2019, 93, e02130-18. [Google Scholar] [CrossRef]
- Legendre, M.; Lartigue, A.; Bertaux, L.; Jeudy, S.; Bartoli, J.; Lescot, M.; Alempic, J.-M.; Ramus, C.; Bruley, C.; Labadie, K.; et al. In-depth study of Mollivirus sibericum, a new 30,000-y-old giant virus infecting Acanthamoeba. Proc. Natl. Acad. Sci. USA 2015, 112, E5327–E5335. [Google Scholar] [CrossRef]
- Legendre, M.; Bartoli, J.; Shmakova, L.; Jeudy, S.; Labadie, K.; Adrait, A.; Lescot, M.; Poirot, O.; Bertaux, L.; Bruley, C.; et al. Thirty-thousand-year-old distant relative of giant icosahedral DNA viruses with a pandoravirus morphology. Proc. Natl. Acad. Sci. USA 2014, 111, 4274–4279. [Google Scholar] [CrossRef] [PubMed]
- Risler, A.; Coupat-Goutaland, B.; Pélandakis, M. Genotyping and phylogenetic analysis of Acanthamoeba isolates associated with keratitis. Parasitol. Res. 2013, 112, 3807–3816. [Google Scholar] [CrossRef] [PubMed]
- Maumus, F.; Blanc, G. Study of gene trafficking between acanthamoeba and giant viruses suggests an undiscovered family of amoeba-infecting viruses. Genome Biol. Evol. 2016, 8, 3351–3363. [Google Scholar] [CrossRef] [PubMed]
- Garate, M.; Cubillos, I.; Marchant, J.; Panjwani, N. Biochemical characterization and functional studies of acanthamoeba mannose-binding protein. Infect. Immun. 2005, 73, 5775–5781. [Google Scholar] [CrossRef] [PubMed]
- Panjwani, N. Pathogenesis of acanthamoeba keratitis. Ocul. Surf. 2010, 8, 70–79. [Google Scholar] [CrossRef]
- Hurt, M.; Niederkorn, J.; Alizadeh, H. Effects of mannose on Acanthamoeba castellanii proliferation and cytolytic ability to corneal epithelial cells. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3424–3431. [Google Scholar] [CrossRef] [PubMed]
- Garate, M.; Alizadeh, H.; Neelam, S.; Niederkorn, J.Y.; Panjwani, N. Oral immunization with acanthamoeba castellanii mannose-binding protein ameliorates amoebic keratitis. Infect. Immun. 2006, 74, 7032–7034. [Google Scholar] [CrossRef]
- Mortazavi, P.N.; Keisary, E.; Loh, L.N.; Jung, S.-Y.; Khan, N.A. Possible roles of phospholipase A2 in the biological activities of acanthamoeba castellanii (T4 genotype). Protist 2011, 162, 168–176. [Google Scholar] [CrossRef]
- Pérez-Serrano, J.; Martínez, J.; Pérez, B.; Bernadina, W.E.; Rodríguez-Caabeiro, F. In vitro shock response to different stressors in free living and pathogenic Acanthamoeba. Int. J. Parasitol. 2000, 30, 829–835. [Google Scholar] [CrossRef]
- Podlipaeva, I.I.; Shmakov, L.A.; Gilichinskiĭ, D.A.; Gudkov, A.V. Heat shock protein of HSP70 family revealed in some contemporary freshwater Amoebae and in Acanthamoeba sp. from cysts isolated from permafrost samples. Tsitologiia 2006, 48, 691–694. [Google Scholar]
- Cirillo, J.D.; Falkow, S.; Tompkins, L.S. Growth of legionella pneumophila in acanthamoeba castellanii enhances invasion. Infect. Immun. 1994, 62, 3254–3261. [Google Scholar] [CrossRef] [PubMed]
- Hasni, I.; Chelkha, N.; Baptiste, E.; Mameri, M.R.; Lachuer, J.; Plasson, F.; Colson, P.; Scola, B.L. Investigation of potential pathogenicity of Willaertia magna by investigating the transfer of bacteria pathogenicity genes into its genome. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- La Scola, B.; Audic, S.; Robert, C.; Jungang, L.; de Lamballerie, X.; Drancourt, M.; Birtles, R.; Claverie, J.-M.; Raoult, D. A giant virus in amoebae. Science 2003, 299, 2033. [Google Scholar] [CrossRef]
- Andreani, J.; Khalil, J.Y.B.; Sevvana, M.; Benamar, S.; Di Pinto, F.; Bitam, I.; Colson, P.; Klose, T.; Rossmann, M.G.; Raoult, D.; et al. Pacmanvirus, a new giant icosahedral virus at the crossroads between asfarviridae and faustoviruses. J. Virol. 2017, 91, e00212-17. [Google Scholar] [CrossRef] [PubMed]
- Colson, P.; Pagnier, I.; Yoosuf, N.; Fournous, G.; La Scola, B.; Raoult, D. “Marseilleviridae”, a new family of giant viruses infecting amoebae. Arch. Virol. 2013, 158, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Philippe, N.; Legendre, M.; Doutre, G.; Couté, Y.; Poirot, O.; Lescot, M.; Arslan, D.; Seltzer, V.; Bertaux, L.; Bruley, C.; et al. Pandoraviruses: Amoeba viruses with genomes up to 2.5 mb reaching that of parasitic eukaryotes. Science 2013, 341, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Khalil, J.Y.B.; Andreani, J.; La Scola, B. Updating strategies for isolating and discovering giant viruses. Curr. Opin. Microbiol. 2016, 31, 80–87. [Google Scholar] [CrossRef]
- Abrahão, J.; Silva, L.; Silva, L.S.; Khalil, J.Y.B.; Rodrigues, R.; Arantes, T.; Assis, F.; Boratto, P.; Andrade, M.; Kroon, E.G.; et al. Tailed giant Tupanvirus possesses the most complete translational apparatus of the known virosphere. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Niyyati, M.; Abedkhojasteh, H.; Salehi, M.; Farnia, S.; Rezaeian, M. Axenic cultivation and pathogenic assays of acanthamoeba strains using physical parameters. Iran. J. Parasitol. 2013, 8, 186–189. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Altschul, S.F. BLAST Algorithm. In eLS.; American Cancer Society: Atlanta, GA, USA, 2014; ISBN 978-0-470-01590-2. [Google Scholar]
- Nadalin, F.; Vezzi, F.; Policriti, A. GapFiller: A de novo assembly approach to fill the gap within paired reads. BMC Bioinform. 2012, 13, S8. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Stanke, M.; Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33, W465–W467. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef]
- Jensen, L.J.; Julien, P.; Kuhn, M.; von Mering, C.; Muller, J.; Doerks, T.; Bork, P. eggNOG: Automated construction and annotation of orthologous groups of genes. Nucleic Acids Res. 2008, 36, D250–D254. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Hong, J.; Ji, J.; Xu, J.; Cao, W.; Liu, Z.; Sun, X. An unusual case of Acanthamoeba Polyphaga and Pseudomonas Aeruginosa keratitis. Diagn. Pathol. 2014, 9, 105. [Google Scholar] [CrossRef]
- González-Robles, A.; Omaña-Molina, M.; Salazar-Villatoro, L.; Flores-Maldonado, C.; Lorenzo-Morales, J.; Reyes-Batlle, M.; Arnalich-Montiel, F.; Martínez-Palomo, A. Acanthamoeba culbertsoni isolated from a clinical case with intraocular dissemination: Structure and in vitro analysis of the interaction with hamster cornea and MDCK epithelial cell monolayers. Exp. Parasitol. 2017, 183, 245–253. [Google Scholar] [CrossRef]
- Ledee, D.R.; Hay, J.; Byers, T.J.; Seal, D.V.; Kirkness, C.M. Acanthamoeba griffini. Molecular characterization of a new corneal pathogen. Investig. Ophthalmol. Vis. Sci. 1996, 37, 544–550. [Google Scholar]
- Arnalich-Montiel, F.; Lumbreras-Fernández, B.; Martín-Navarro, C.M.; Valladares, B.; Lopez-Velez, R.; Morcillo-Laiz, R.; Lorenzo-Morales, J. Influence of acanthamoeba genotype on clinical course and outcomes for patients with acanthamoeba keratitis in Spain. J. Clin. Microbiol. 2014, 52, 1213–1216. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing Large Minimum Evolution Trees with Profiles instead of a Distance Matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef]
- Lechner, M.; Findeiß, S.; Steiner, L.; Marz, M.; Stadler, P.F.; Prohaska, S.J. Proteinortho: Detection of (Co-)orthologs in large-scale analysis. BMC Bioinform. 2011, 12, 124. [Google Scholar] [CrossRef]
- Contreras-Moreira, B.; Vinuesa, P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 2013, 79, 7696–7701. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Number |
|---|---|
| Haploid genome size (bp) | 66,434,030 |
| Sequence contigs (n) | 13,849 |
| GC-content (%) | 58.6 |
| Maximal scaffold size (bp) | 80,033 |
| Minimal scaffold size (bp) | 980 |
| Average scaffold size | 5322 |
| N50 | 8852 |
| N75 | 4376 |
| Organisms | Genome Size (Mb) | Predicted Proteins | Annotated Proteins | G+C % |
|---|---|---|---|---|
| Acanthamoeba triangularis ATCC 50254 | 66 | 37,062 | 33,168 | 58.6 |
| Acanthamoeba castellanii ATCC 50370 | 42 | 20,681 | 15,455 | 57.8 |
| Willaertia magna C2c Maky | 37 | 18,519 | 13,571 | 25 |
| Naegleria fowleri ATCC 30863 | 30 | 17,252 | 16,021 | 35 |
| Naegleria gruberi NEG-M | 41 | 15,727 | 9090 | 33 |
| Naegleria lovaniensis ATCC 30569 | 31 | 15,195 | 13,005 | 37 |
| Dictyostelium discoideum AX4 | 34 | 13,541 | 8422 | 22 |
| Entamoeba histolytica strain HM-1: IMSS | 21 | 8201 | 4076 | 24 |
| Gene Identification | Function |
|---|---|
| Adhesion | |
| gene 34934 | mannose binding |
| Metalloproteases | |
| gene 12288 | Aminopeptidase I zinc metalloprotease (M18) |
| gene 1047 | metalloenzyme superfamily |
| gene 9969 | metallocarboxypeptidase |
| gene 3258 | metalloenzyme |
| Proteases | |
| gene 19397 | peptidase S8 and S53 subtilisin kexin sedolisin |
| gene 3757 | PFAM peptidase T2 asparaginase 2 |
| gene 8789 | peptidase C19 family |
| gene 9969 | metallocarboxypeptidase |
| gene 11390 | peptidase S8 and S53, subtilisin, kexin, sedolisin |
| gene 12288 | Aminopeptidase I zinc metalloprotease (M18) |
| gene 12995 | peptidase M17 family |
| gene 25924 | Serine aminopeptidase, S33 |
| gene 26649 | peptidase C19 family |
| gene 27916 | peptidase C19 family |
| gene 9026 | peptidase C19 family protein |
| Temperature tolerance | |
| gene 6737 | Hsp20/alpha crystallin family |
| gene 1586 | Hsp70 protein |
| gene 8645 | Hsp20/alpha crystallin family protein |
| Phospholipases | |
| gene 8337 | phospholipase A2 activator activity |
| gene 27752 | phospholipase D |
| gene 34693 | phospholipase D |
| Antioxidant defense | |
| gene 15075 | glutathione peroxidase |
| gene 31764 | peroxidase |
| gene 27487 | oxidoreductase |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasni, I.; Andréani, J.; Colson, P.; La Scola, B. Description of Virulent Factors and Horizontal Gene Transfers of Keratitis-Associated Amoeba Acanthamoeba Triangularis by Genome Analysis. Pathogens 2020, 9, 217. https://doi.org/10.3390/pathogens9030217
Hasni I, Andréani J, Colson P, La Scola B. Description of Virulent Factors and Horizontal Gene Transfers of Keratitis-Associated Amoeba Acanthamoeba Triangularis by Genome Analysis. Pathogens. 2020; 9(3):217. https://doi.org/10.3390/pathogens9030217
Chicago/Turabian StyleHasni, Issam, Julien Andréani, Philippe Colson, and Bernard La Scola. 2020. "Description of Virulent Factors and Horizontal Gene Transfers of Keratitis-Associated Amoeba Acanthamoeba Triangularis by Genome Analysis" Pathogens 9, no. 3: 217. https://doi.org/10.3390/pathogens9030217
APA StyleHasni, I., Andréani, J., Colson, P., & La Scola, B. (2020). Description of Virulent Factors and Horizontal Gene Transfers of Keratitis-Associated Amoeba Acanthamoeba Triangularis by Genome Analysis. Pathogens, 9(3), 217. https://doi.org/10.3390/pathogens9030217