Establishment of a Plasmid-Based Reverse Genetics System for the Cell Culture-Adapted Hepatitis E Virus Genotype 3c Strain 47832c

 and
and 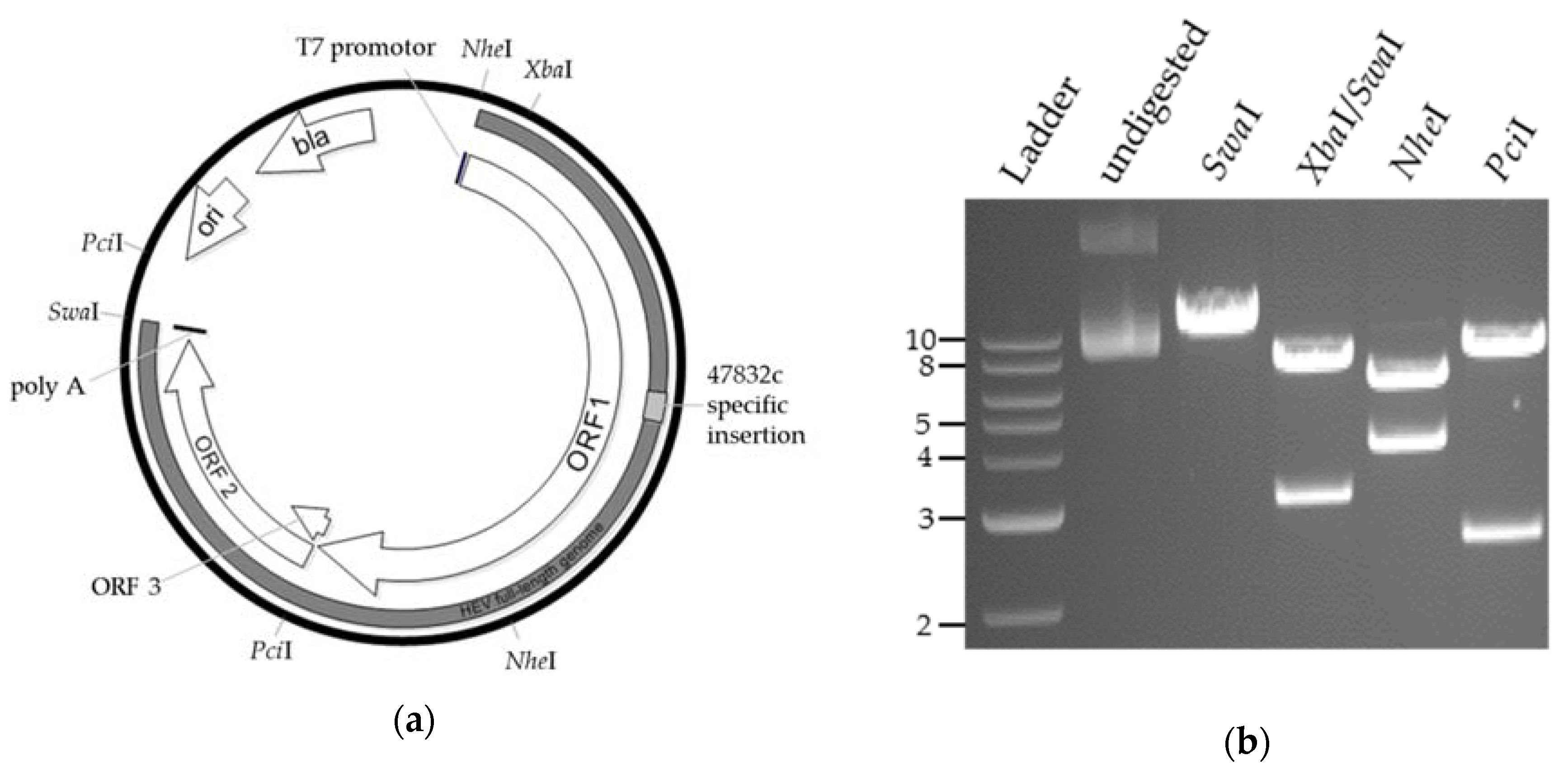{kind=link}
{kind=link}
{kind=link}
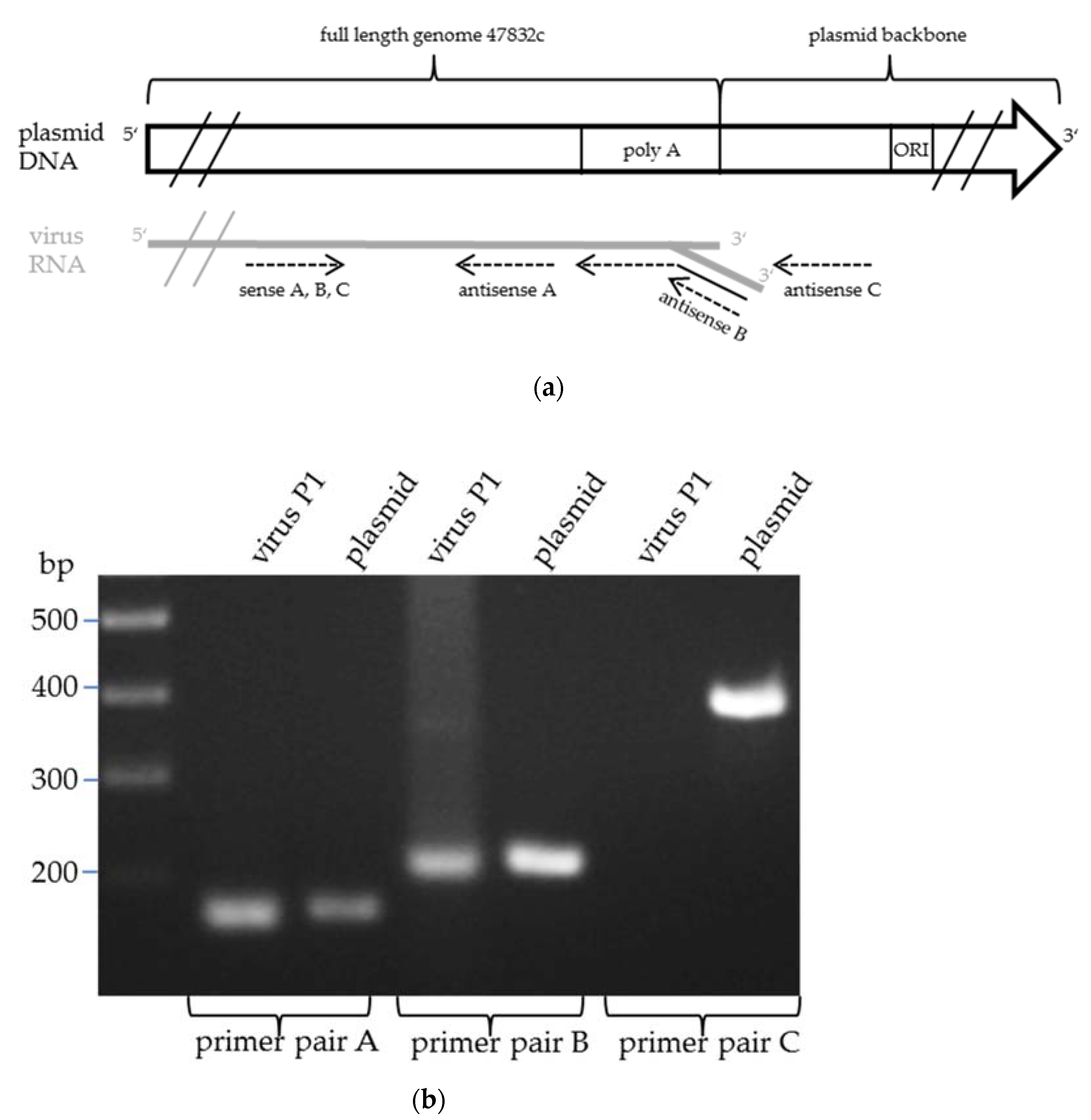{kind=link}
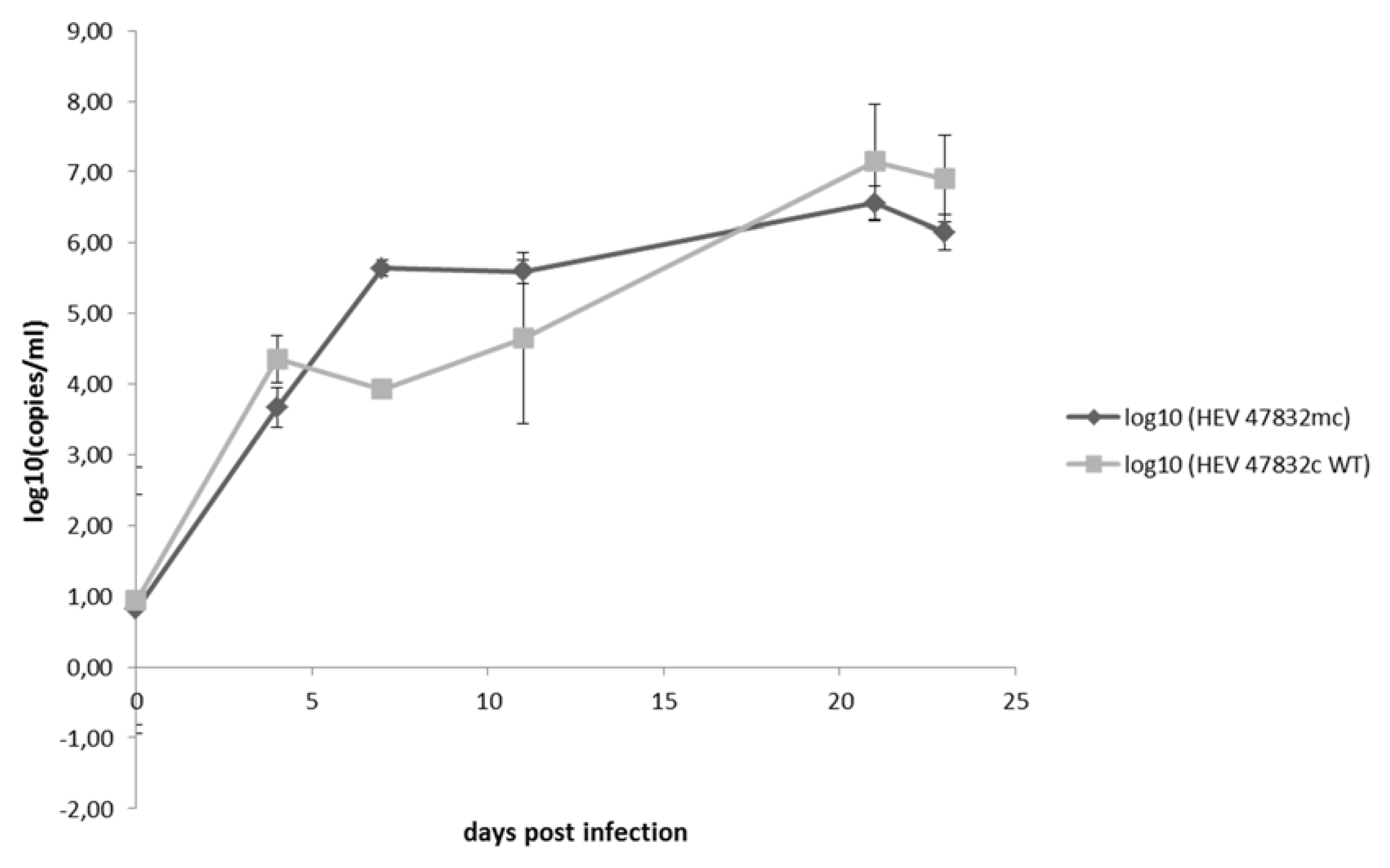{kind=link}
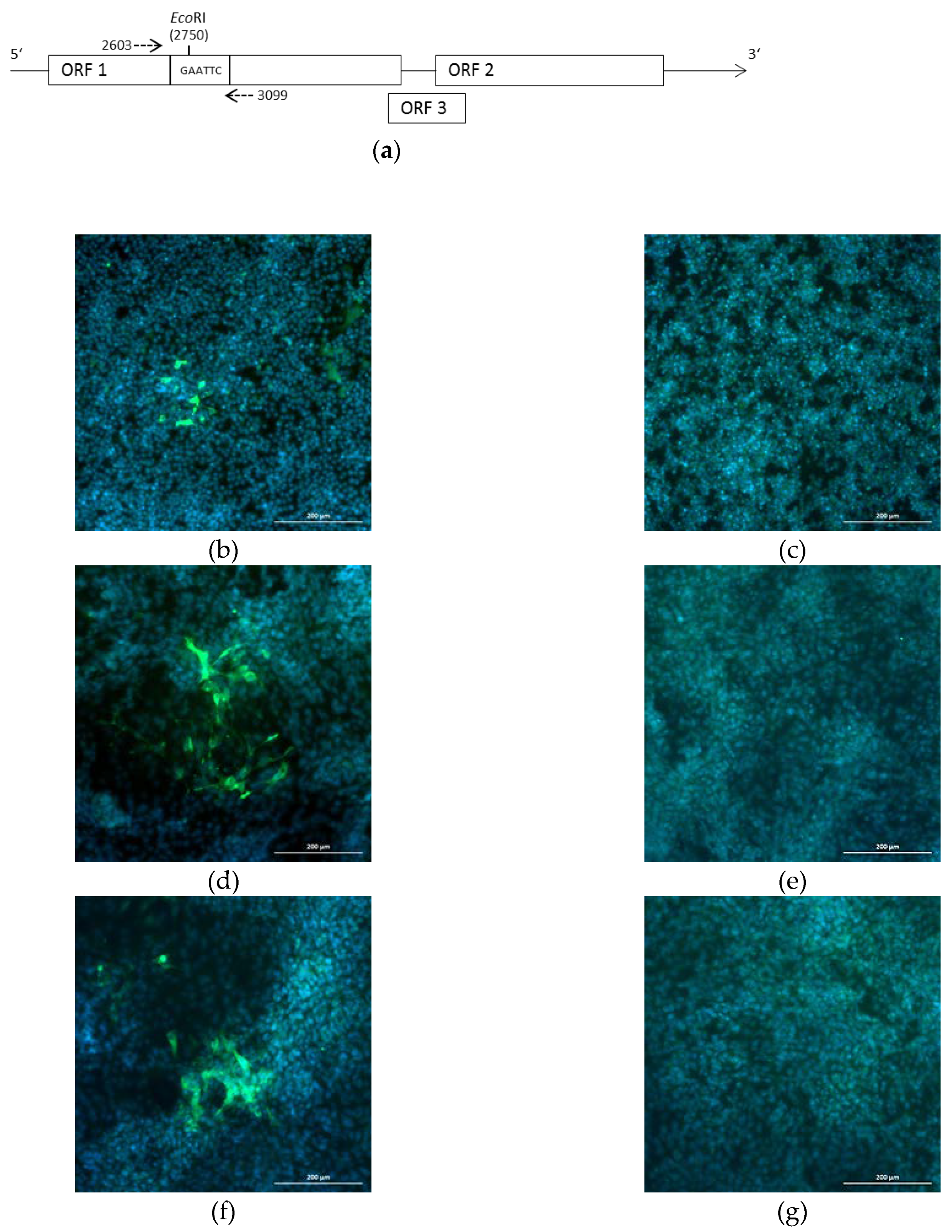{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
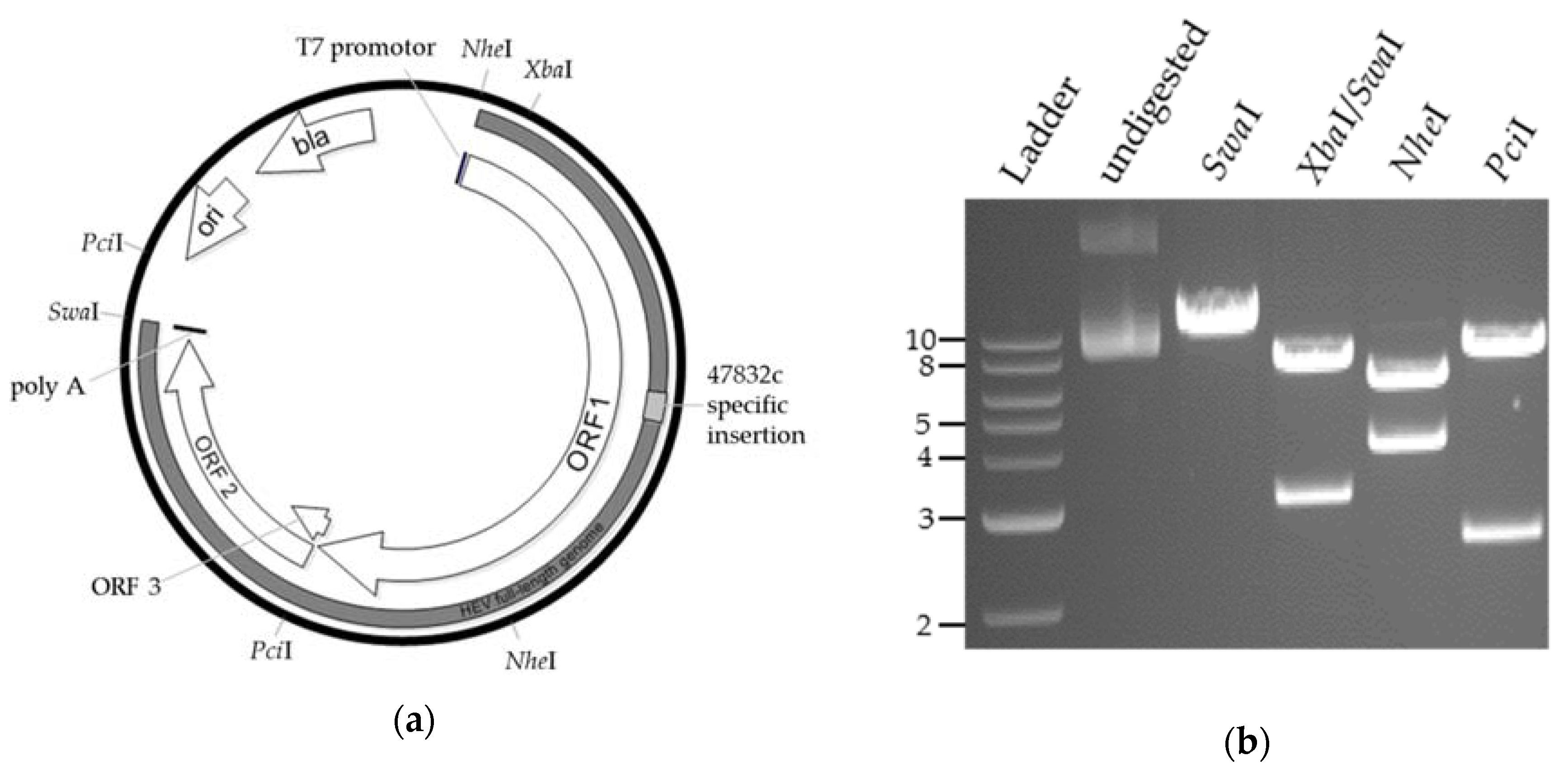
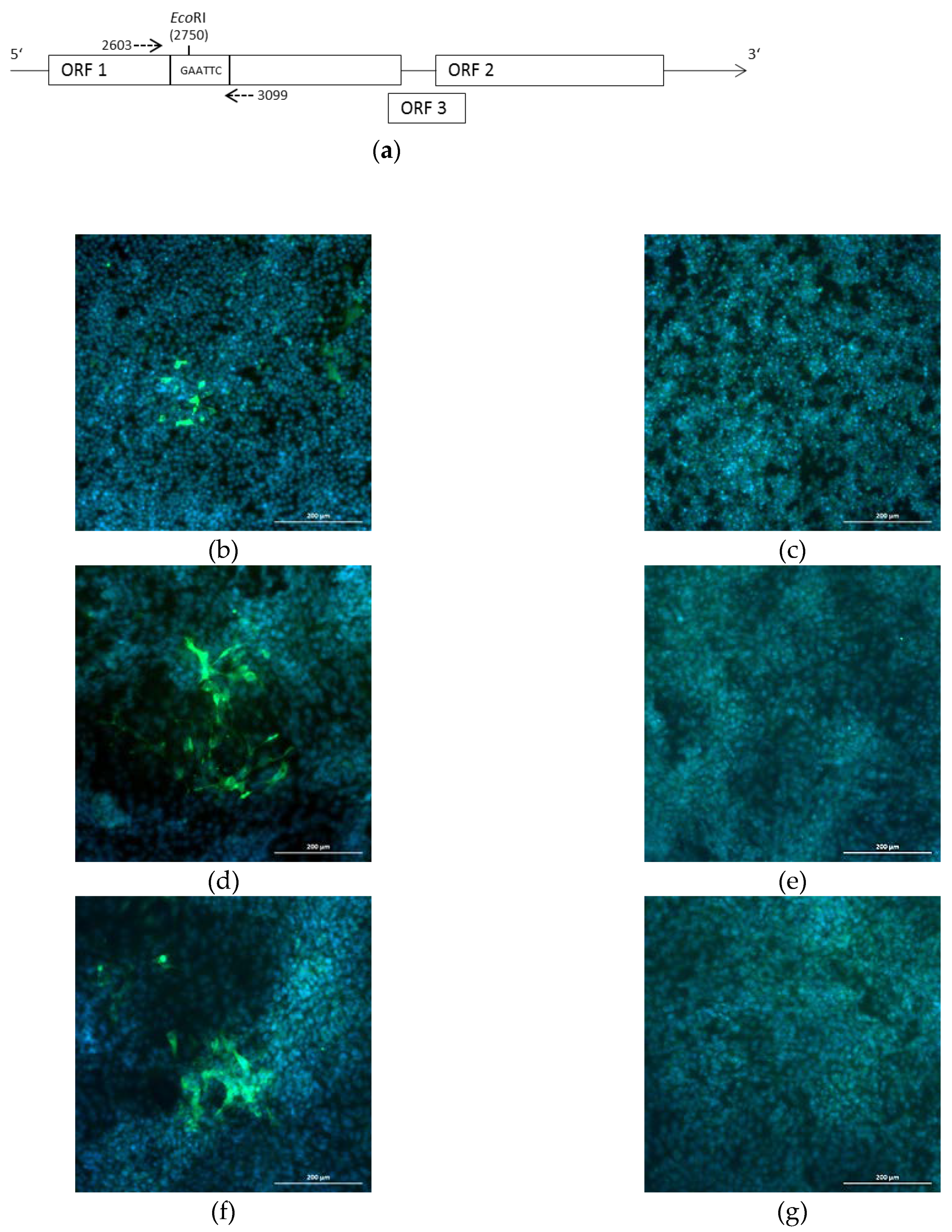
2.1. Generation of a Genomic Plasmid of the HEV Strain 47832c
2.2. Generation of Infectious HEV by Transfection of the Genomic Plasmid
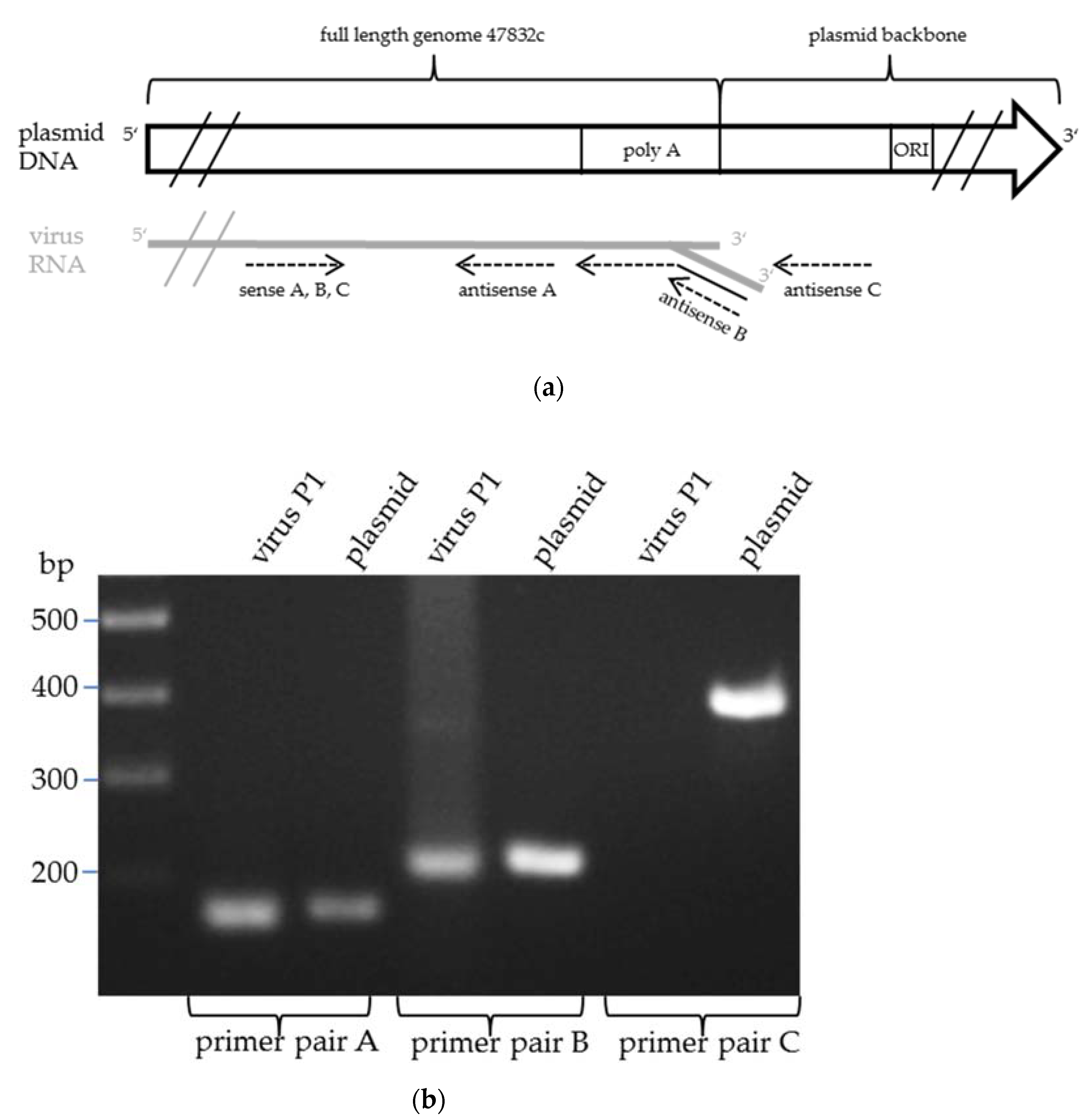
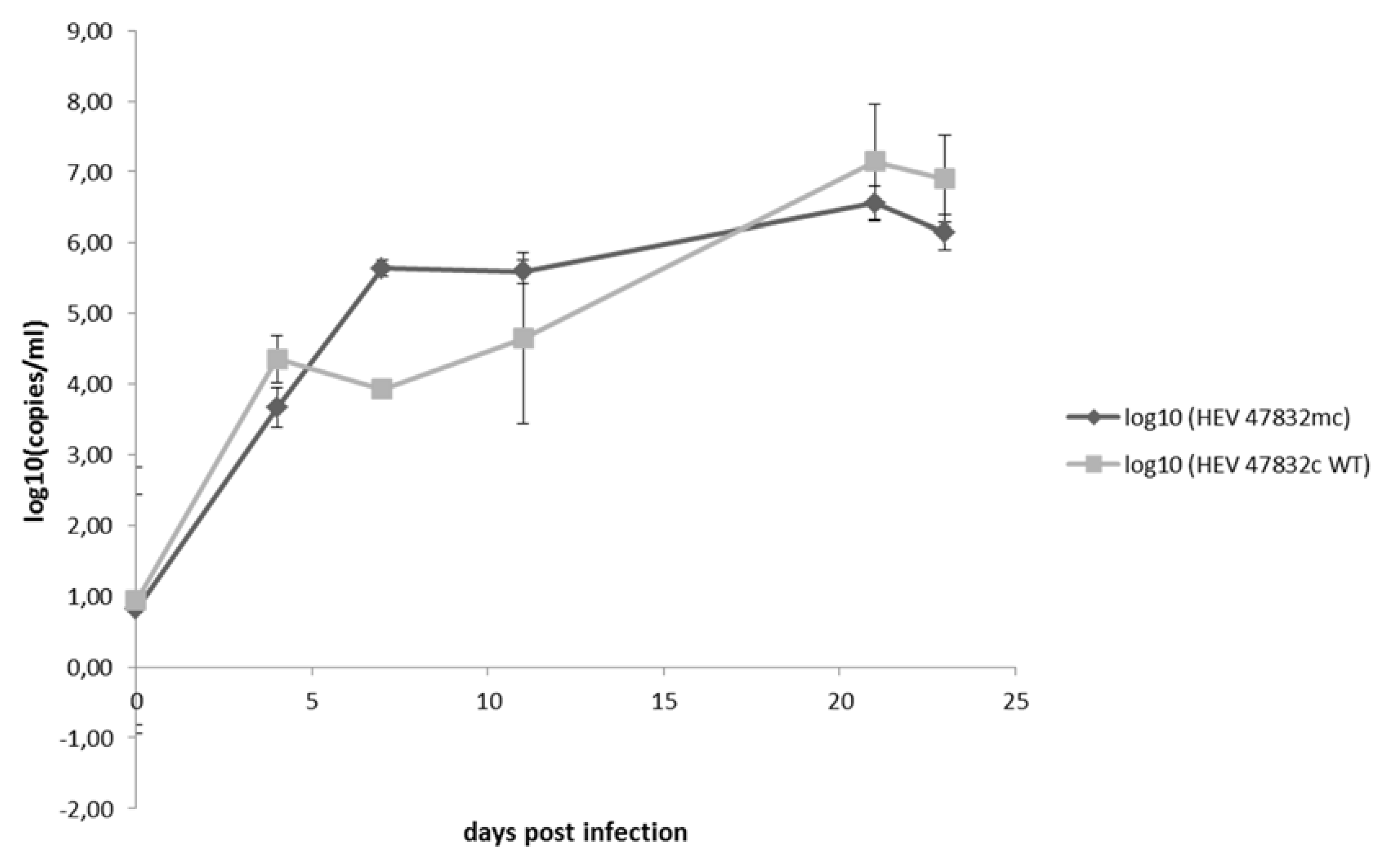
2.3. Characterization of the Generated Virus
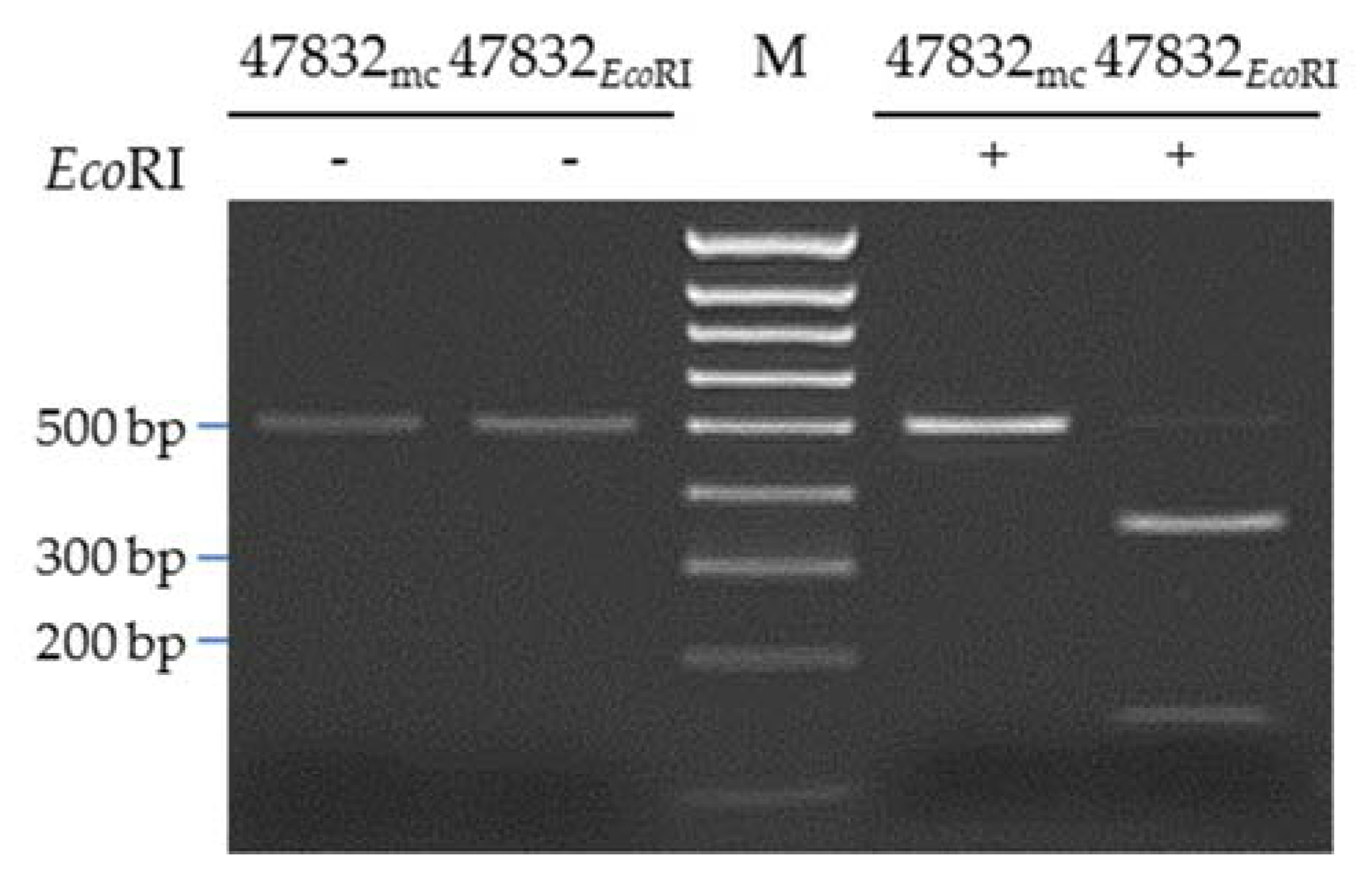
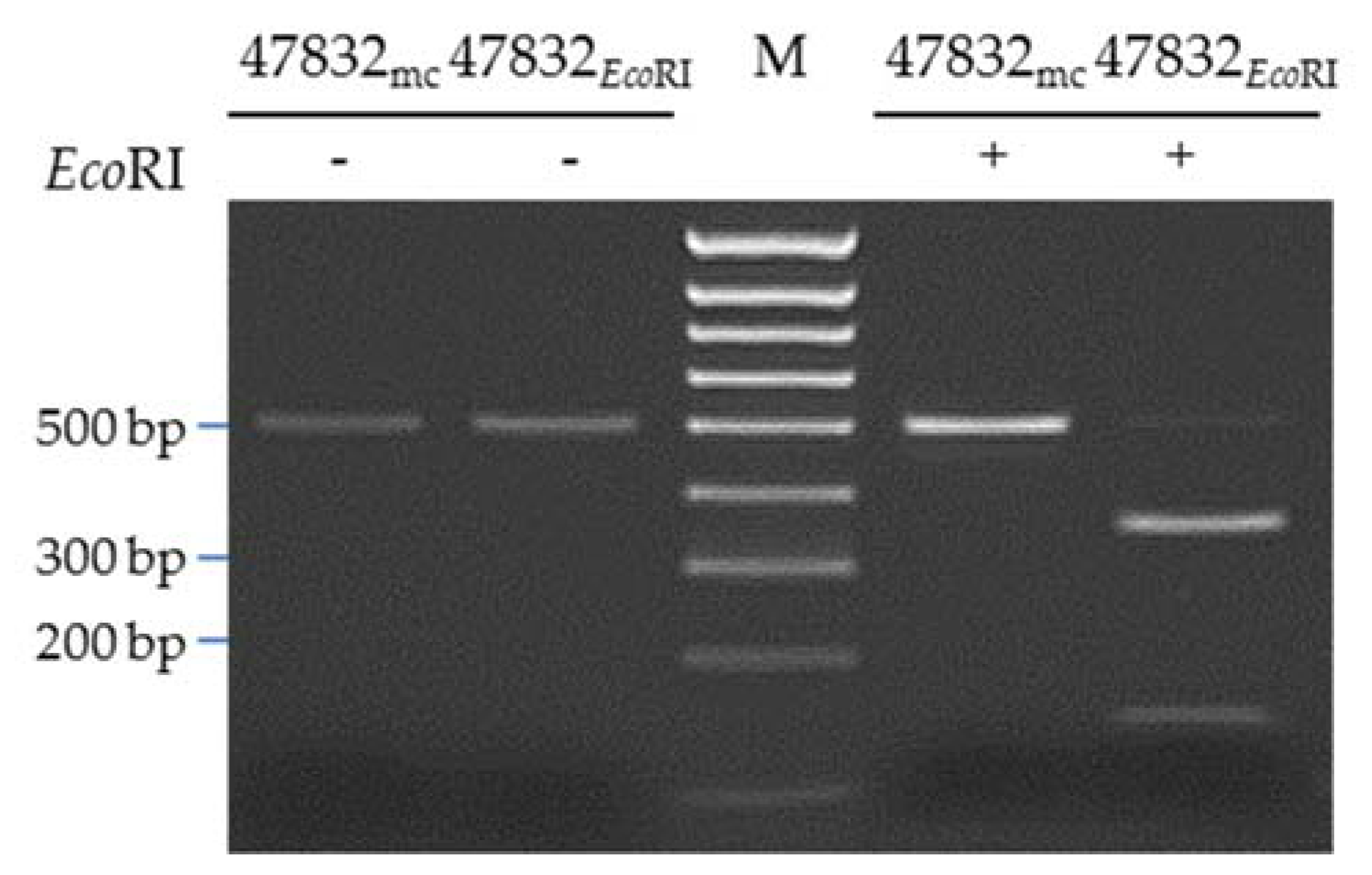
2.4. Introduction of a Point Mutation into the Virus Genome
3. Discussion
4. Materials and Methods
4.1. Cells and Viruses
4.2. Generation of the Genomic Plasmid p47832mc
4.3. Generation of the Mutated Plasmid p47832EcoRI
4.4. Generation of Infectious Virus from Plasmids
4.5. Immunofluorescence Analysis
4.6. Electron Microscopy
4.7. Growth Kinetics of Viruses
4.8. PCR Analysis of the 3′-end of the Virus Genome
4.9. PCR and Restriction Fragment Analysis of Mutant 47832EcoRI
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Fact sheet Hepatitis E. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-e (accessed on 8 July 2019).
- Khuroo, M.S.; Khuroo, M.S. Hepatitis E: An emerging global disease—from discovery towards control and cure. J. Viral Hep. 2016, 23, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Aspinall, E.J.; Couturier, E.; Faber, M.; Said, B.; Ijaz, S.; Tavoschi, L.; Takkinen, L.; Adlhoch, C. Hepatitis E virus infection in Europe: Surveillance and descriptive epidemiology of confirmed cases, 2005 to 2015. Eurosurveillance 2017, 22, 30561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purdy, M.A.; Harrison, T.J.; Jameel, S.; Meng, X.J.; Okamoto, H.; Van der Poel, W.H.M.; Smith, D.B. ICTV Report Consortium. ICTV Virus Taxonomy Profile: Hepeviridae. J. Gen. Virol. 2017, 98, 2645–2646. [Google Scholar] [PubMed]
- Nagashima, S.; Takahashi, M.; Kobayashi, T.; Tanggis; Nishizawa, T.; Nishiyama, T.; Primadharsini, P.P.; Okamoto, H. Characterization of the Quasi-Enveloped Hepatitis E Virus Particles Released by the Cellular Exosomal Pathway. J. Virol. 2017, 91, e00822-17. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.B.; Simmonds, P.; Izopet, J.; Oliveira-Filho, E.F.; Ulrich, R.G.; Johne, R.; Koenig, M.; Jameel, S.; Harrison, T.J.; Meng, X.-J.; et al. Proposed reference sequences for hepatitis E virus subtypes. J. Gen. Virol. 2016, 97, 537–542. [Google Scholar] [CrossRef]
- Pavio, N.; Doceul, V.; Bagdassarian, E.; Johne, R. Recent knowledge on hepatitis E virus in Suidae reservoirs and transmission routes to human. Vet. Res. 2017, 48, 78. [Google Scholar] [CrossRef] [Green Version]
- Meister, T.L.; Bruening, J.; Todt, D.; Steinmann, E. Cell culture systems for the study of hepatitis E virus. Antiviral Res. 2019, 163, 34–49. [Google Scholar] [CrossRef]
- Schemmerer, M.; Johne, R.; Erl, M.; Jilg, W.; Wenzel, J.J. Isolation of Subtype 3c, 3e and 3f-like Hepatitis E Virus Strains Stably Replicating to High Viral Loads in an Optimized Cell Culture System. Viruses 2019, 11, 483. [Google Scholar] [CrossRef] [Green Version]
- Shukla, P.; Nguyen, H.T.; Faulk, K.; Mather, K.; Torian, U.; Engle, R.E.; Emerson, S.U. Adaptation of a genotype 3 hepatitis E virus to efficient growth in cell culture depends on an inserted human gene segment acquired by recombination. J. Virol. 2012, 86, 5697–5707. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.; Takahashi, M.; Hoshino, Y.; Takahashi, H.; Ichiyama, K.; Tanaka, T.; Okamoto, H. Construction of an infectious cDNA clone of hepatitis E virus strain JE03-1760F that can propagate efficiently in cultured cells. J. Gen. Virol. 2009, 90, 457–462. [Google Scholar] [CrossRef]
- Adlhoch, C.; Avellon, A.; Baylis, S.A.; Ciccaglione, A.R.; Couturier, E.; de Sousa, R.; Epstein, J.; Ethelberg, S.; Faber, M.; Feher, A.; et al. Hepatitis E virus: Assessment of the epidemiological situation in humans in Europe, 2014/15. J. Clin. Virol. 2016, 82, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Johne, R.; Reetz, J.; Ulrich, R.G.; Machnowska, P.; Sachsenroder, J.; Nickel, P.; Hofmann, J. An ORF1-rearranged hepatitis E virus derived from a chronically infected patient efficiently replicates in cell culture. J. Viral Hep. 2014, 21, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Schemmerer, M.; Apelt, S.; Trojnar, E.; Ulrich, R.G.; Wenzel, J.J.; Johne, R. Enhanced Replication of Hepatitis E Virus Strain 47832c in an A549-Derived Subclonal Cell Line. Viruses 2016, 8, 267. [Google Scholar] [CrossRef] [PubMed]
- Johne, R.; Trojnar, E.; Filter, M.; Hofmann, J. Thermal stability of hepatitis E virus estimated by a cell culture method. Appl. Environ. Microbiol. 2016, 82, 4225–4231. [Google Scholar] [CrossRef] [Green Version]
- Glitscher, M.; Himmelsbach, K.; Woytinek, K.; Johne, R.; Reuter, A.; Spiric, J.; Schwaben, L.; Grünweller, A.; Hildt, E. Inhibition of Hepatitis E virus spread by the natural compound silvestrol. Viruses 2018, 10, 301. [Google Scholar] [CrossRef] [Green Version]
- Shukla, P.; Nguyen, H.T.; Torian, U.; Engle, R.E.; Faulk, K.; Dalton, H.R.; Bendall, R.P.; Keane, F.E.; Purcell, R.H.; Emerson, S.U. Cross-species infections of cultured cells by hepatitis E virus and discovery of an infectious virus-host recombinant. Proc. Natl. Acad. Sci. USA 2011, 108, 2438–2443. [Google Scholar] [CrossRef] [Green Version]
- Emerson, S.U.; Zhang, M.; Meng, X.J.; Nguyen, H.; St Claire, M.; Govindarajan, S.; Huang, Y.K.; Purcell, R.H. Recombinant hepatitis E virus genomes infectious for primates: importance of capping and discovery of a cis-reactive element. Proc. Natl. Acad. Sci. USA 2001, 98, 15270–15275. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.W.; Haqshenas, G.; Kasorndorkbua, C.; Halbur, P.G.; Emerson, S.U.; Meng, X.J. Capped RNA transcripts of full-length cDNA clones of swine hepatitis E virus are replication competent when transfected into Huh7 cells and infectious when intrahepatically inoculated into pigs. J. Virol. 2005, 79, 1552–1558. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.F.; Pierson, F.W.; Toth, T.E.; Meng, X.J. Construction and characterization of infectious cDNA clones of a chicken strain of hepatitis E virus (HEV), avian HEV. J. Gen. Virol. 2005, 86, 2585–2593. [Google Scholar] [CrossRef]
- Kanai, Y.; Komoto, S.; Kawagishi, T.; Nouda, R.; Nagasawa, N.; Onishi, M.; Matsuura, Y.; Taniguchi, K.; Kobayashi, T. Entirely plasmid-based reverse genetics system for rotaviruses. Proc. Natl. Acad. Sci. USA 2017, 114, 2349–2354. [Google Scholar] [CrossRef] [Green Version]
- Debing, Y.; Gisa, A.; Pischke, S.; Bremer, B.; Manns, M.; Wedemeyer, H.; Suneetha, P.V.; Neyts, J. A mutation in the hepatitis E virus RNA polymerase promotes its replication and associates with ribavirin treatment failure in organ transplant recipients. Gastroenterology 2014, 147, 1008–1011. [Google Scholar]
- Todt, D.; Friesland, M.; Moeller, N.; Praditya, D.; Kinast, V.; Brüggemann, Y.; Knegendorf, L.; Burkard, T.; Steinmann, J.; Burm, R.; et al. Robust hepatitis E virus infection and transcriptional response in human hepatocytes. Proc. Natl. Acad. Sci. USA 2020, 117, 1731–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchholz, U.J.; Finke, S.; Conzelmann, K.K. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J. Virol. 1999, 73, 251–259. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jothikumar, N.; Cromeans, T.L.; Robertson, B.H.; Meng, X.J.; Hill, V.R. A broadly reactive one-step real-time RT-PCR assay for rapid and sensitive detection of hepatitis E virus. J. Virol. Methods 2006, 131, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Schielke, A.; Filter, M.; Appel, B.; Johne, R. Thermal stability of hepatitis E virus assessed by a molecular biological approach. Virol. J. 2011, 8, 487. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scholz, J.; Bächlein, C.; Gadicherla, A.K.; Falkenhagen, A.; Tausch, S.H.; Johne, R. Establishment of a Plasmid-Based Reverse Genetics System for the Cell Culture-Adapted Hepatitis E Virus Genotype 3c Strain 47832c. Pathogens 2020, 9, 157. https://doi.org/10.3390/pathogens9030157
Scholz J, Bächlein C, Gadicherla AK, Falkenhagen A, Tausch SH, Johne R. Establishment of a Plasmid-Based Reverse Genetics System for the Cell Culture-Adapted Hepatitis E Virus Genotype 3c Strain 47832c. Pathogens. 2020; 9(3):157. https://doi.org/10.3390/pathogens9030157
Chicago/Turabian StyleScholz, Johannes, Christine Bächlein, Ashish K. Gadicherla, Alexander Falkenhagen, Simon H. Tausch, and Reimar Johne. 2020. "Establishment of a Plasmid-Based Reverse Genetics System for the Cell Culture-Adapted Hepatitis E Virus Genotype 3c Strain 47832c" Pathogens 9, no. 3: 157. https://doi.org/10.3390/pathogens9030157
APA StyleScholz, J., Bächlein, C., Gadicherla, A. K., Falkenhagen, A., Tausch, S. H., & Johne, R. (2020). Establishment of a Plasmid-Based Reverse Genetics System for the Cell Culture-Adapted Hepatitis E Virus Genotype 3c Strain 47832c. Pathogens, 9(3), 157. https://doi.org/10.3390/pathogens9030157