Map3k14 as a Regulator of Innate and Adaptive Immune Response during Acute Viral Infection


 , , ,
, , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Genome-Wide Association Study Shows That Map3k14 Is a Regulator of Viral Replication in the Spleen
2.2. Map3k14 Promotes Innate and Adaptive Immune Activation
2.3. Map3k14 Signaling Maintains Splenic Marginal Zone Architecture
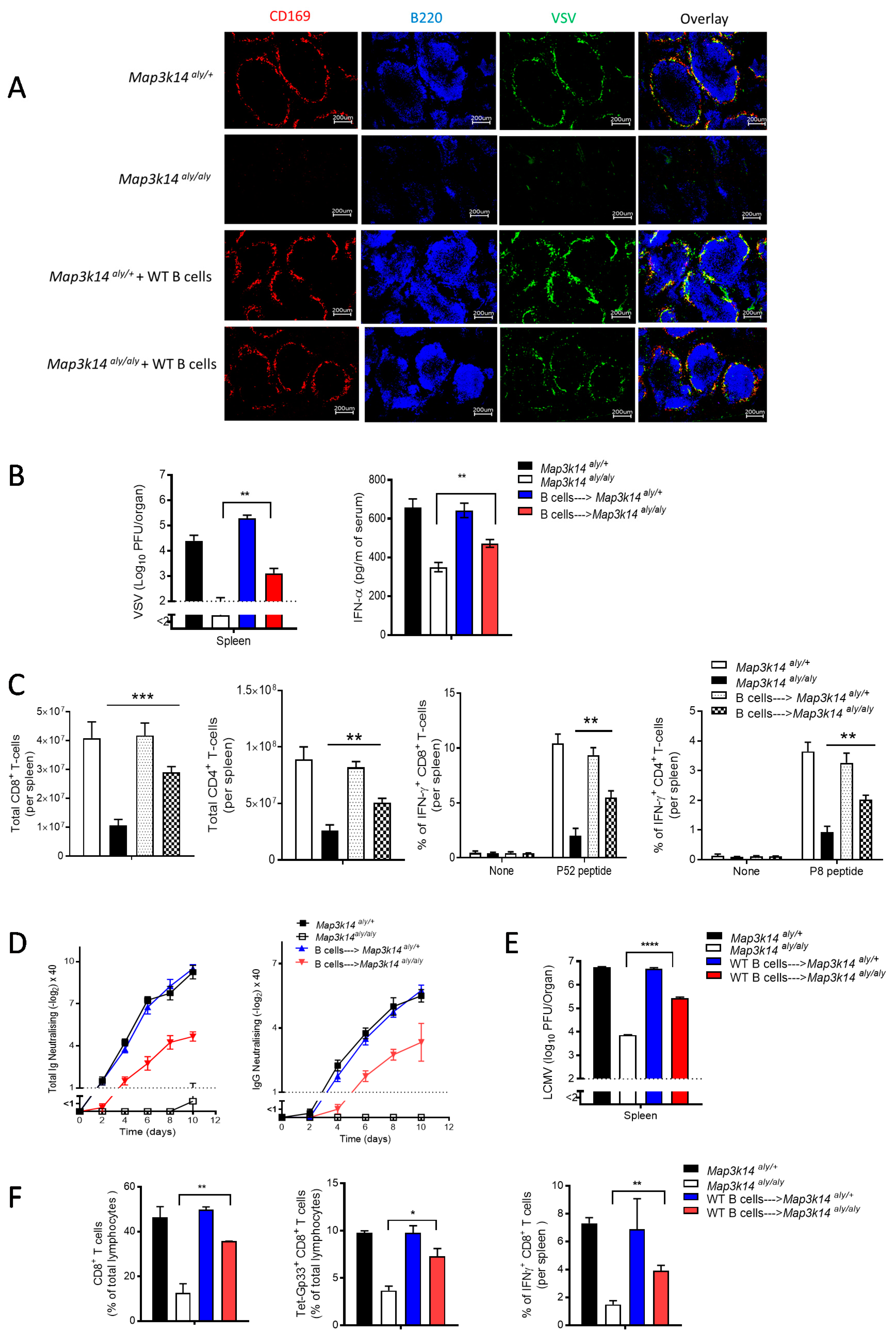
2.4. B Cells Are Required for the Development and Immune Activation of Splenic CD169 Macrophages in a Map3k14 Dysfunctional State
3. Discussion
4. Methods
4.1. Mice
4.2. Genome-Wide Association Study
4.3. Viruses
4.4. Bone Marrow Chimeras
4.5. Plaque Assay
4.6. Neutralizing Antibody Assay
4.7. Purification and Adoptive Transfer of B Cells
4.8. Tetramer, Surface and Intracellular Staining, and Flow Cytometry
4.9. Cell Preparation and Staining for Dendritic Cell Subsets Study
4.10. Histology
4.11. ELISA
4.12. Statistical analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Randall, T.D.; Carragher, D.M.; Rangel-Moreno, J. Development of secondary lymphoid organs. Annu. Rev. Immunol. 2008, 26, 627–650. [Google Scholar] [CrossRef]
- Lewis, S.M.; Williams, A.; Eisenbarth, S.C. Structure and function of the immune system in the spleen. Sci. Immunol. 2019, 4, eaau6085. [Google Scholar] [CrossRef] [PubMed]
- Mebius, R.E.; Kraal, G. Structure and function of the spleen. Nat. Rev. Immunol. 2005, 5, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, A.; Cols, M.; Puga, I. Marginal zone B cells: Virtues of innate-like antibody-producing lymphocytes. Nat. Rev. Immunol. 2013, 13, 118–132. [Google Scholar] [CrossRef]
- Bendelac, A.; Bonneville, M.; Kearney, J.F. Autoreactivity by design: Innate B and T lymphocytes. Nat. Rev. Immunol. 2001, 1, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Zouali, M.; Richard, Y. Marginal zone B-cells, a gatekeeper of innate immunity. Front. Immunol. 2011, 2, 63. [Google Scholar] [CrossRef] [PubMed]
- Belperron, A.A.; Dailey, C.M.; Booth, C.J.; Bockenstedt, L.K. Marginal zone B-cell depletion impairs murine host defense against borrelia burgdorferi infection. Infect. Immun. 2007, 75, 3354–3360. [Google Scholar] [CrossRef] [PubMed]
- Junt, T.; Moseman, E.A.; Iannacone, M.; Massberg, S.; Lang, P.A.; Boes, M.; Fink, K.; Henrickson, S.E.; Shayakhmetov, D.M.; Mempel, T.R.; et al. Subcapsular sinus macrophages in lymph nodes clear lymph-borne viruses and present them to antiviral B cells. Nature 2007, 450, 110–114. [Google Scholar] [CrossRef]
- Gretz, J.E.; Anderson, A.O.; Shaw, S. Cords, channels, corridors and conduits: Critical architectural elements facilitating cell interactions in the lymph node cortex. Immunol. Rev. 1997, 156, 11–24. [Google Scholar] [CrossRef]
- Sixt, M.; Kanazawa, N.; Selg, M.; Samson, T.; Roos, G.; Reinhardt, D.P.; Pabst, R.; Lutz, M.B.; Sorokin, L. The conduit system transports soluble antigens from the afferent lymph to resident dendritic cells in the T cell area of the lymph node. Immunity 2005, 22, 19–29. [Google Scholar] [CrossRef]
- Oehen, S.; Odermatt, B.; Karrer, U.; Hengartner, H.; Zinkernagel, R.; Lopez-Macias, C. Marginal zone macrophages and immune responses against viruses. J. Immunol. 2002, 169, 1453–1458. [Google Scholar] [CrossRef] [PubMed]
- Seiler, P.; Aichele, P.; Odermatt, B.; Hengartner, H.; Zinkernagel, R.M.; Schwendener, R.A. Crucial role of marginal zone macrophages and marginal zone metallophils in the clearance of lymphocytic choriomeningitis virus infection. Eur. J. Immunol. 1997, 27, 2626–2633. [Google Scholar] [CrossRef] [PubMed]
- Honke, N.; Shaabani, N.; Cadeddu, G.; Sorg, U.R.; Zhang, D.E.; Trilling, M.; Klingel, K.; Sauter, M.; Kandolf, R.; Gailus, N.; et al. Enforced viral replication activates adaptive immunity and is essential for the control of a cytopathic virus. Nat. Immunol. 2011, 13, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Pikkarainen, T.; Elomaa, O.; Soininen, R.; Kodama, T.; Kraal, G.; Karl, T. Defective microarchitecture of the spleen marginal zone and impaired response to a thymus-independent type 2 antigen in mice lacking scavenger receptors MARCO and SR-A. J. Immunol. 2005, 175, 8173–8180. [Google Scholar] [CrossRef] [PubMed]
- Dingjan, G.M.; Maas, A.; Nawijn, M.C.; Smit, L.; Voerman, J.S.; Grosveld, F.; Hendriks, R.W. Severe B cell deficiency and disrupted splenic architecture in transgenic mice expressing the E41K mutated form of Bruton’s tyrosine kinase. EMBO J. 1998, 17, 5309–5320. [Google Scholar] [CrossRef] [PubMed]
- Nolte, M.A.; Arens, R.; Kraus, M.; van Oers, M.H.; Kraal, G.; van Lier, R.A.; Mebius, R.E. B cells are crucial for both development and maintenance of the splenic marginal zone. J. Immunol. 2004, 172, 3620–3627. [Google Scholar] [CrossRef]
- Karlsson, M.C.; Guinamard, R.; Bolland, S.; Sankala, M.; Steinman, R.M.; Ravetch, J.V. Macrophages control the retention and trafficking of B lymphocytes in the splenic marginal zone. J. Exp. Med. 2003, 198, 333–340. [Google Scholar] [CrossRef]
- Shinkura, R.; Kitada, K.; Matsuda, F.; Tashiro, K.; Ikuta, K.; Suzuki, M.; Kogishi, K.; Serikawa, T.; Honjo, T. Alymphoplasia is caused by a point mutation in the mouse gene encoding Nf-kappa b-inducing kinase. Nat. Genet. 1999, 22, 74–77. [Google Scholar] [CrossRef]
- Miyawaki, S.; Nakamura, Y.; Suzuka, H.; Koba, M.; Yasumizu, R.; Ikehara, S. A new mutation, aly, that induces a generalized lack of lymph nodes accompanied by immunodeficiency in mice. Eur. J. Immunol. 1994, 24, 429–434. [Google Scholar] [CrossRef]
- Koike, R.; Nishimura, T.; Yasumizu, R.; Tanaka, H.; Hataba, Y.; Hataba, Y.; Watanabe, T.; Miyawaki, S.; Miyasaka, M. The splenic marginal zone is absent in alymphoplastic aly mutant mice. Eur. J. Immunol. 1996, 26, 669–675. [Google Scholar] [CrossRef]
- Karrer, U.; Althage, A.; Odermatt, B.; Hengartner, H.; Zinkernagel, R.M. Immunodeficiency of alymphoplasia mice (aly/aly) in vivo: Structural defect of secondary lymphoid organs and functional B cell defect. Eur. J. Immunol. 2000, 30, 2799–2807. [Google Scholar] [CrossRef]
- Hahn, M.; Macht, A.; Waisman, A.; Hovelmeyer, N. NF-kappaB-inducing kinase is essential for B-cell maintenance in mice. Eur. J. Immunol. 2016, 46, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Kirby, A.; Kang, H.M.; Wade, C.M.; Cotsapas, C.; Kostem, E.; Han, B.; Furlotte, N.; Kang, E.Y.; Rivas, M.; Bogue, M.A.; et al. Fine mapping in 94 inbred mouse strains using a high-density haplotype resource. Genetics 2010, 185, 1081–1095. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Zaitlen, N.A.; Wade, C.M.; Kirby, A.; Heckerman, D.; Daly, M.J.; Eskin, E. Efficient control of population structure in model organism association mapping. Genetics 2008, 178, 1709–1723. [Google Scholar] [CrossRef]
- Klein, A.D.; Ferreira, N.S.; Ben-Dor, S.; Duan, J.; Hardy, J.; Cox, T.M.; Merrill, A.H., Jr.; Futerman, A.H. Identification of modifier genes in a mouse model of gaucher Disease. Cell Rep. 2016, 16, 2546–2553. [Google Scholar] [CrossRef]
- Honke, N.; Shaabani, N.; Zhang, D.E.; Iliakis, G.; Xu, H.C.; Haussinger, D.; Recher, M.; Löhning, M.; Lang, K.S.; Lang, K.S. Usp18 driven enforced viral replication in dendritic cells contributes to break of immunological tolerance in autoimmune diabetes. PLoS Pathog. 2013, 9, e1003650. [Google Scholar] [CrossRef]
- Duhan, V.; Khairnar, V.; Friedrich, S.K.; Zhou, F.; Gassa, A.; Honke, N.; Shaabani, N.; Gailus, N.; Botezatu, L.; Khandanpour, C.; et al. Virus-specific antibodies allow viral replication in the marginal zone, thereby promoting CD8(+) T-cell priming and viral control. Sci. Rep. 2016, 6, 19191. [Google Scholar] [CrossRef]
- Minor, P.D. Live attenuated vaccines: Historical successes and current challenges. Virology 2015, 479-480, 379–392. [Google Scholar] [CrossRef]
- Yu, J.; Zhou, X.; Nakaya, M.; Jin, W.; Cheng, X.; Sun, S.C. T cell-intrinsic function of the noncanonical NF-kappaB pathway in the regulation of GM-CSF expression and experimental autoimmune encephalomyelitis pathogenesis. J. Immunol. 2014, 193, 422–430. [Google Scholar] [CrossRef]
- Karrer, U.; Althage, A.; Odermatt, B.; Roberts, C.W.; Korsmeyer, S.J.; Miyawaki, S.; Hengartner, H.; Zinkernagel, R.M. On the key role of secondary lymphoid organs in antiviral immune responses studied in alymphoplastic (aly/aly) and spleenless (Hox11(-)/-) mutant mice. J. Exp. Med. 1997, 185, 2157–2170. [Google Scholar] [CrossRef]
- Tumanov, A.; Kuprash, D.; Lagarkova, M.; Grivennikov, S.; Abe, K.; Shakhov, A.N.; Drutskaya, L.N.; Stewart, C.L.; Chervonsky, A.V.; Nedospasov, S.A. Distinct role of surface lymphotoxin expressed by B cells in the organization of secondary lymphoid tissues. Immunity 2002, 17, 239–250. [Google Scholar] [CrossRef]
- Xu, H.C.; Huang, J.; Khairnar, V.; Duhan, V.; Pandyra, A.A.; Grusdat, M.; Shinde, P.; Mcllwain, D.R.; Maney, S.K.; Gommerman, J.; et al. Deficiency of the B cell-activating factor receptor results in limited CD169+ macrophage function during viral infection. J. Virol. 2015, 89, 4748–4759. [Google Scholar] [CrossRef]
- Futterer, A.; Mink, K.; Luz, A.; Kosco-Vilbois, M.H.; Pfeffer, K. The lymphotoxin beta receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity 1998, 9, 59–70. [Google Scholar] [CrossRef]
- Garceau, N.; Kosaka, Y.; Masters, S.; Hambor, J.; Shinkura, R.; Honjo, T.; Noelle, R.J. Lineage-restricted function of nuclear factor kappaB-inducing kinase (NIK) in transducing signals via CD40. J. Exp. Med. 2000, 191, 381–386. [Google Scholar] [CrossRef]
- Benedict, C.A.; Banks, T.A.; Senderowicz, L.; Ko, M.; Britt, W.J.; Angulo, A.; Ghazal, P.; Ware, C.F. Lymphotoxins and cytomegalovirus cooperatively induce interferon-beta, establishing host-virus detente. Immunity 2001, 15, 617–626. [Google Scholar] [CrossRef]
- Schneider, K.; Loewendorf, A.; De Trez, C.; Fulton, J.; Rhode, A.; Shumway, H.; Ha, S.; Patterson, G.; Pfeffer, K.; Ware, C.F.; et al. Lymphotoxin-mediated crosstalk between B cells and splenic stroma promotes the initial type I interferon response to cytomegalovirus. Cell Host Microbe 2008, 3, 67–76. [Google Scholar] [CrossRef]
- Fink, K.; Lang, K.S.; Manjarrez-Orduno, N.; Junt, T.; Senn, B.M.; Holdener, M.; Akira, S.; Zinkernagel, R.M.; Hengartner, H. Early type I interferon-mediated signals on B cells specifically enhance antiviral humoral responses. Eur. J. Immunol. 2006, 36, 2094–2105. [Google Scholar] [CrossRef]
- Honke, N.; Shaabani, N.; Merches, K.; Gassa, A.; Kraft, A.; Ehrhardt, K.; Häussinger, D.; Lang, P.A.; Lang, K.S.; Recher, M.; et al. Immunoactivation induced by chronic viral infection inhibits viral replication and drives immunosuppression through sustained IFN-I responses. Eur. J. Immunol. 2016, 46, 372–380. [Google Scholar] [CrossRef]
- Battegay, M.; Cooper, S.; Althage, A.; Banziger, J.; Hengartner, H.; Zinkernagel, R.M. Quantification of lymphocytic choriomeningitis virus with an immunological focus assay in 24-or 96-well plates. J. Virol. Methods 1991, 33, 191–198. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamdan, T.A.; Bhat, H.; Cham, L.B.; Adomati, T.; Lang, J.; Li, F.; Murtaza, A.; Hardt, C.; Lang, P.A.; Duhan, V.; et al. Map3k14 as a Regulator of Innate and Adaptive Immune Response during Acute Viral Infection. Pathogens 2020, 9, 96. https://doi.org/10.3390/pathogens9020096
Hamdan TA, Bhat H, Cham LB, Adomati T, Lang J, Li F, Murtaza A, Hardt C, Lang PA, Duhan V, et al. Map3k14 as a Regulator of Innate and Adaptive Immune Response during Acute Viral Infection. Pathogens. 2020; 9(2):96. https://doi.org/10.3390/pathogens9020096
Chicago/Turabian StyleHamdan, Thamer A., Hilal Bhat, Lamin B. Cham, Tom Adomati, Judith Lang, Fanghui Li, Ali Murtaza, Cornelia Hardt, Philipp A. Lang, Vikas Duhan, and et al. 2020. "Map3k14 as a Regulator of Innate and Adaptive Immune Response during Acute Viral Infection" Pathogens 9, no. 2: 96. https://doi.org/10.3390/pathogens9020096
APA StyleHamdan, T. A., Bhat, H., Cham, L. B., Adomati, T., Lang, J., Li, F., Murtaza, A., Hardt, C., Lang, P. A., Duhan, V., & Lang, K. S. (2020). Map3k14 as a Regulator of Innate and Adaptive Immune Response during Acute Viral Infection. Pathogens, 9(2), 96. https://doi.org/10.3390/pathogens9020096