Computational Analysis of African Swine Fever Virus Protein Space for the Design of an Epitope-Based Vaccine Ensemble

 ,
,  ,
,  and
and
Abstract
1. Introduction
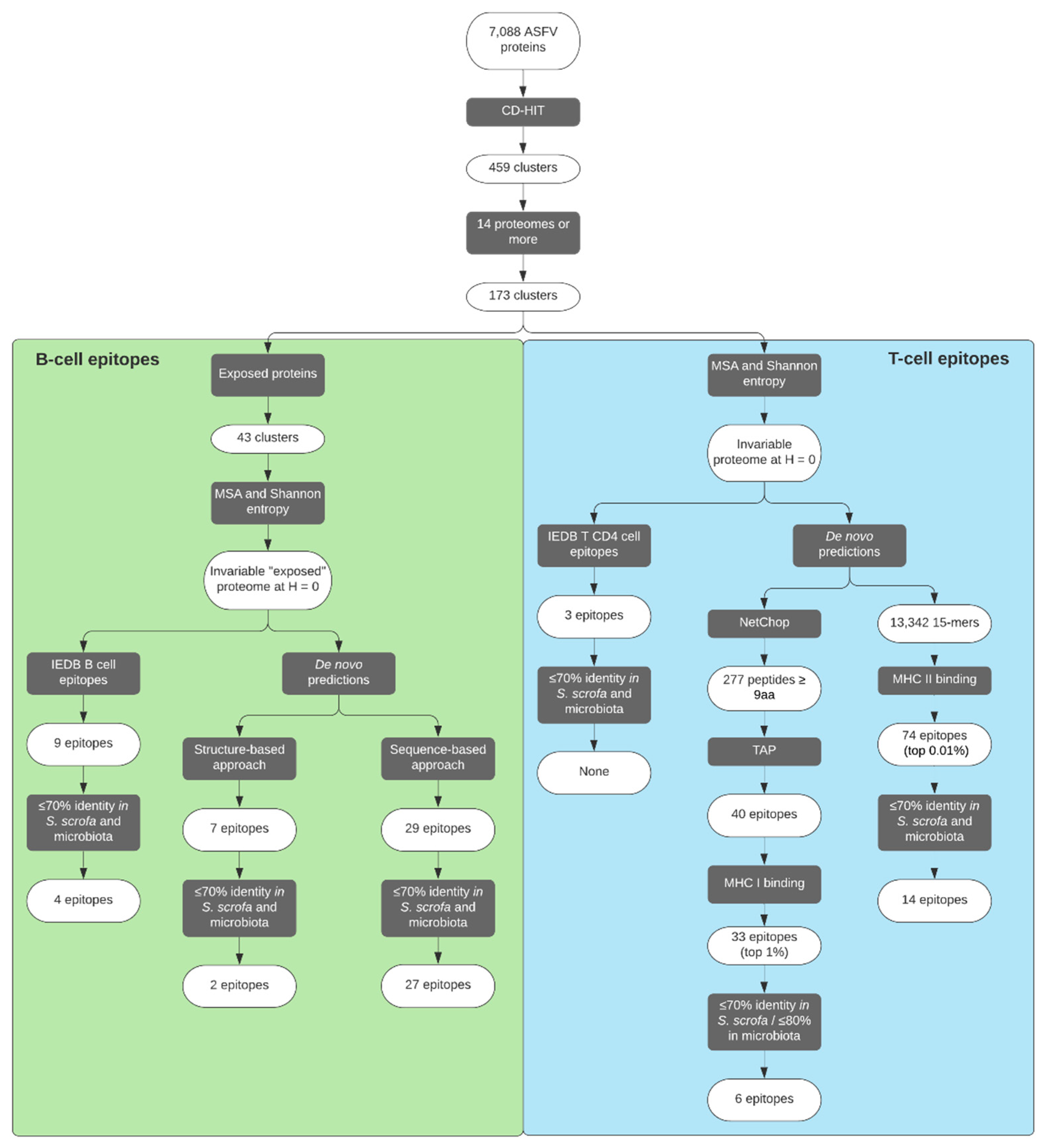
2. Results
2.1. Analysis of ASFV Protein Space
2.2. Identification of ASFV Putative-Exposed Proteins
2.3. Selection of B-Cell Epitopes
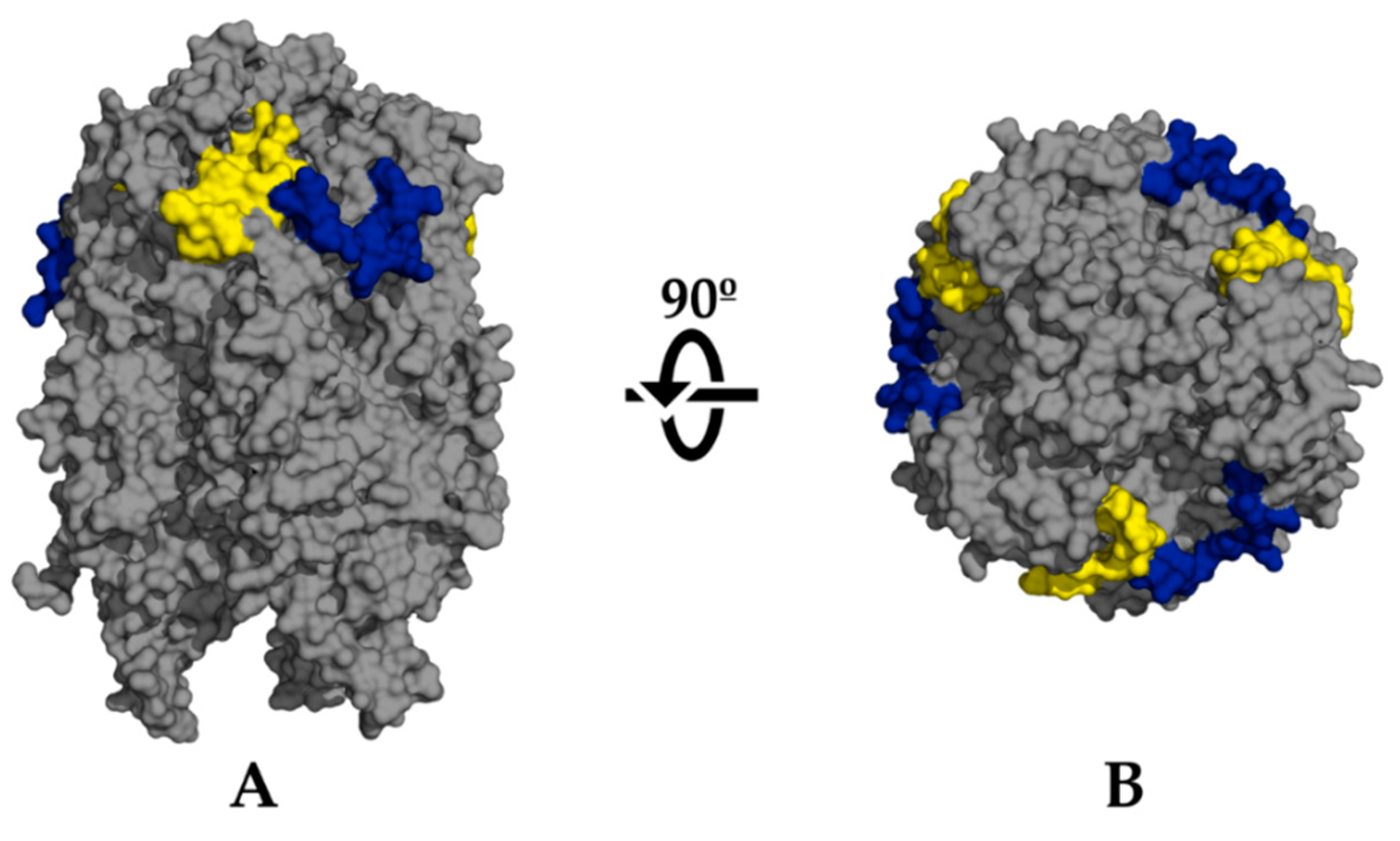
2.4. De Novo Predicted B-Cell Epitopes
2.5. Selection of CD4+ T-Cell Epitopes
2.6. De Novo Predicted CD4+ T-Cell Epitopes
2.7. Selection of CD8+ T-Cell Epitopes
3. Discussion
4. Materials and Methods
4.1. Collection of ASFV Proteomes
4.2. Collection of Experimentally Validated ASFV Epitopes
4.3. Clustering of ASFV Proteins and Generation of Multiple Sequence Alignments
4.4. Generating an ASFV Invariable Proteome
4.5. Prediction of CD8+ T-Cell Epitopes
4.6. Prediction of CD4+ T-Cell Epitopes
4.7. Prediction of B-Cell Epitopes
4.8. In-House Scripts and BLAST Searches
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Montgomery, R.E. On A Form of Swine Fever Occurring in British East Africa (Kenya Colony). J. Comp. Pathol. Ther. 1921, 34, 159–191. [Google Scholar] [CrossRef]
- Cisek, A.A.; Dąbrowska, I.; Gregorczyk, K.P.; Wyżewski, Z. African Swine Fever Virus: A new old enemy of Europe. Ann. Parasitol. 2016, 62, 161–167. [Google Scholar] [CrossRef]
- Dixon, L.K.; Stahl, K.; Jori, F.; Vial, L.; Pfeiffer, D.U. African Swine Fever Epidemiology and Control. Annu. Rev. Anim. Biosci. 2020, 8, 221–246. [Google Scholar] [CrossRef] [PubMed]
- Guinat, C.; Gogin, A.; Blome, S.; Keil, G.; Pollin, R.; Pfeiffer, D.U.; Dixon, L. Transmission routes of African swine fever virus to domestic pigs: Current knowledge and future research directions. Vet. Rec. 2016, 178, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Costard, S.; Wieland, B.; De Glanville, W.; Jori, F.; Rowlands, R.; Vosloo, W.; Roger, F.; Pfeiffer, D.U.; Dixon, L.K. African swine fever: How can global spread be prevented? Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 2683–2696. [Google Scholar] [CrossRef] [PubMed]
- Dixon, L.K.; Sun, H.; Roberts, H. African swine fever. Antivir. Res. 2019, 165, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Luo, Y.; Wang, Y.; Li, S.; Zhao, Z.; Bi, Y.; Sun, J.; Peng, R.; Song, H.; Zhu, D.; et al. Cryo-EM Structure of the African Swine Fever Virus. Cell Host Microbe 2019, 26, 836–843.e3. [Google Scholar] [CrossRef] [PubMed]
- Andrés, G.; Charro, D.; Matamoros, T.; Dillard, R.S.; Abrescia, N.G.A. The cryo-EM structure of African swine fever virus unravels a unique architecture comprising two icosahedral protein capsids and two lipoprotein membranes. J. Biol. Chem. 2020, 295, 1–12. [Google Scholar] [CrossRef]
- Wang, N.; Zhao, D.; Wang, J.J.; Zhang, Y.; Wang, M.; Gao, Y.; Li, F.; Wang, J.J.; Bu, Z.; Rao, Z.; et al. Architecture of African swine fever virus and implications for viral assembly. Science 2019, 366, 640–644. [Google Scholar] [CrossRef]
- Franzoni, G.; Dei Giudici, S.; Oggiano, A. Infection, modulation and responses of antigen-presenting cells to African swine fever viruses. Virus Res. 2018, 258, 73–80. [Google Scholar] [CrossRef]
- Dixon, L.K.; Chapman, D.A.G.; Netherton, C.L.; Upton, C. African swine fever virus replication and genomics. Virus Res. 2013, 173, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Portugal, R.; Coelho, J.; Höper, D.; Little, N.S.; Smithson, C.; Upton, C.; Martins, C.; Leitão, A.; Keil, G.M. Related strains of African swine fever virus with different virulence: Genome comparison and analysis. J. Gen. Virol. 2015, 96, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, J.M.; Moreno, L.T.; Alejo, A.; Lacasta, A.; Rodríguez, F.; Salas, M.L. Genome Sequence of African Swine Fever Virus BA71, the Virulent Parental Strain of the Nonpathogenic and Tissue-Culture Adapted BA71V. PLoS ONE 2015, 10, e0142889. [Google Scholar] [CrossRef] [PubMed]
- Alejo, A.; Matamoros, T.; Guerra, M.; Andrés, G. A Proteomic Atlas of the African Swine Fever Virus Particle. J. Virol. 2018, 92, 1–18. [Google Scholar] [CrossRef]
- Yáñez, R.J.; Rodríguez, J.M.; Nogal, M.L.; Yuste, L.; Enríquez, C.; Rodriguez, J.F.; Viñuela, E. Analysis of the Complete Nucleotide Sequence of African Swine Fever Virus. Virology 1995, 208, 249–278. [Google Scholar] [CrossRef]
- The Uniprot Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Bastos, A.D.S.; Penrith, M.L.; Crucière, C.; Edrich, J.L.; Hutchings, G.; Roger, F.; Couacy-Hymann, E.; Thomson, G.R. Genotyping field strains of African swine fever virus by partial p72 gene characterisation. Arch. Virol. 2003, 148, 693–706. [Google Scholar] [CrossRef]
- Gaudreault, N.N.; Madden, D.W.; Wilson, W.C.; Trujillo, J.D.; Richt, J.A. African Swine Fever Virus: An Emerging DNA Arbovirus. Front. Vet. Sci. 2020, 7, 215. [Google Scholar] [CrossRef]
- Malogolovkin, A.; Burmakina, G.; Titov, I.; Sereda, A.; Gogin, A.; Baryshnikova, E.; Kolbasov, D. Comparative analysis of african swine fever virus genotypes and serogroups. Emerg. Infect. Dis. 2015, 21, 312–315. [Google Scholar] [CrossRef]
- Monteagudo, P.L.; Lacasta, A.; López, E.; Bosch, L.; Collado, J.; Pina-Pedrero, S.; Correa-Fiz, F.; Accensi, F.; Navas, M.J.; Vidal, E.; et al. BA71ΔCD2: A New Recombinant Live Attenuated African Swine Fever Virus with Cross-Protective Capabilities. J. Virol. 2017, 91, 1–17. [Google Scholar] [CrossRef]
- O’Donnell, V.; Risatti, G.R.; Holinka, L.G.; Krug, P.W.; Carlson, J.; Velazquez-Salinas, L.; Azzinaro, P.A.; Gladue, D.P.; Borca, M.V. Simultaneous Deletion of the 9GL and UK Genes from the African Swine Fever Virus Georgia 2007 Isolate Offers Increased Safety and Protection against Homologous Challenge. J. Virol. 2017, 91, e01760-16. [Google Scholar] [CrossRef] [PubMed]
- Revilla, Y.; Pérez-Núñez, D.; Richt, J.A. African Swine Fever Virus Biology and Vaccine Approaches. Adv. Virus Res. 2018, 100, 41–74. [Google Scholar] [CrossRef] [PubMed]
- Borca, M.V.; Ramirez-Medina, E.; Silva, E.; Vuono, E.; Rai, A.; Pruitt, S.; Holinka, L.G.; Velazquez-Salinas, L.; Zhu, J.; Gladue, D.P. Development of a Highly Effective African Swine Fever Virus Vaccine by Deletion of the I177L Gene Results in Sterile Immunity against the Current Epidemic Eurasia Strain. J. Virol. 2020, 94, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhao, D.; He, X.; Liu, R.; Wang, Z.; Zhang, X.; Li, F.; Shan, D.; Chen, H.; Zhang, J.; et al. A seven-gene-deleted African swine fever virus is safe and effective as a live attenuated vaccine in pigs. Sci. China Life Sci. 2020, 63, 623–634. [Google Scholar] [CrossRef]
- Neilan, J.; Zsak, L.; Lu, Z.; Burrage, T.; Kutish, G.; Rock, D. Neutralizing antibodies to African swine fever virus proteins p30, p54, and p72 are not sufficient for antibody-mediated protection. Virology 2004, 319, 337–342. [Google Scholar] [CrossRef]
- Argilaguet, J.M.; Pérez-Martín, E.; Nofrarías, M.; Gallardo, C.; Accensi, F.; Lacasta, A.; Mora, M.; Ballester, M.; Galindo-Cardiel, I.; López-Soria, S.; et al. DNA Vaccination Partially Protects against African Swine Fever Virus Lethal Challenge in the Absence of Antibodies. PLoS ONE 2012, 7, e40942. [Google Scholar] [CrossRef]
- Lacasta, A.; Ballester, M.; Monteagudo, P.L.; Rodriguez, J.M.; Salas, M.L.; Accensi, F.; Pina-Pedrero, S.; Bensaid, A.; Argilaguet, J.; Lopez-Soria, S.; et al. Expression Library Immunization Can Confer Protection against Lethal Challenge with African Swine Fever Virus. J. Virol. 2014, 88, 13322–13332. [Google Scholar] [CrossRef]
- Jancovich, J.K.; Chapman, D.; Hansen, D.T.; Robida, M.D.; Loskutov, A.; Craciunescu, F.; Borovkov, A.; Kibler, K.; Goatley, L.; King, K.; et al. Immunization of Pigs by DNA Prime and Recombinant Vaccinia Virus Boost to Identify and Rank African Swine Fever Virus Immunogenic and Protective Proteins. J. Virol. 2018, 92, e02219-17. [Google Scholar] [CrossRef]
- Lokhandwala, S.; Petrovan, V.; Popescu, L.; Sangewar, N.; Elijah, C.; Stoian, A.; Olcha, M.; Ennen, L.; Bray, J.; Bishop, R.P.; et al. Adenovirus-vectored African Swine Fever Virus antigen cocktails are immunogenic but not protective against intranasal challenge with Georgia 2007/1 isolate. Vet. Microbiol. 2019, 235, 10–20. [Google Scholar] [CrossRef]
- Sang, H.; Miller, G.; Lokhandwala, S.; Sangewar, N.; Waghela, S.D.; Bishop, R.P.; Mwangi, W. Progress Toward Development of Effective and Safe African Swine Fever Virus Vaccines. Front. Vet. Sci. 2020, 7, 1–9. [Google Scholar] [CrossRef]
- Oura, C.A.L.; Denyer, M.S.; Takamatsu, H.; Parkhouse, R.M.E. In vivo depletion of CD8+ T lymphocytes abrogates protective immunity to African swine fever virus. J. Gen. Virol. 2005, 86, 2445–2450. [Google Scholar] [CrossRef] [PubMed]
- Takamatsu, H.-H.; Denyer, M.S.; Lacasta, A.; Stirling, C.M.A.; Argilaguet, J.M.; Netherton, C.L.; Oura, C.A.L.; Martins, C.; Rodríguez, F. Cellular immunity in ASFV responses. Virus Res. 2013, 173, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Netherton, C.L.; Goatley, L.C.; Reis, A.L.; Portugal, R.; Nash, R.H.; Morgan, S.B.; Gault, L.; Nieto, R.; Norlin, V.; Gallardo, C.; et al. Identification and Immunogenicity of African Swine Fever Virus Antigens. Front. Immunol. 2019, 10, 1318. [Google Scholar] [CrossRef] [PubMed]
- Kollnberger, S.D.; Gutierrez-Castañeda, B.; Foster-Cuevas, M.; Corteyn, A.; Parkhouse, R.M.E. Identification of the principal serological immunodeterminants of African swine fever virus by screening a virus cDNA library with antibody. J. Gen. Virol. 2002, 83, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Reis, A.L.; Parkhouse, R.M.E.; Penedos, A.R.; Martins, C.; Leitão, A. Systematic analysis of longitudinal serological responses of pigs infected experimentally with African swine fever virus. J. Gen. Virol. 2007, 88, 2426–2434. [Google Scholar] [CrossRef]
- Gallardo, C.; Mwaengo, D.M.; MacHaria, J.M.; Arias, M.; Taracha, E.A.; Soler, A.; Okoth, E.; Martín, E.; Kasiti, J.; Bishop, R.P. Enhanced discrimination of African swine fever virus isolates through nucleotide sequencing of the p54, p72, and pB602L (CVR) genes. Virus Genes 2009, 38, 85–95. [Google Scholar] [CrossRef]
- Wardley, R.C.; Norley, S.G.; Wilkinson, P.J.; Williams, S. The role of antibody in protection against African swine fever virus. Vet. Immunol. Immunopathol. 1985, 9, 201–212. [Google Scholar] [CrossRef]
- Onisk, D.V.; Borca, M.V.; Kutish, S.; Kramer, E.; Irusta, P.; Rock, D.L. Passively Transferred African Swine Fever Virus Antibodies Protect Swine against Lethal Infection. Virology 1994, 198, 350–354. [Google Scholar] [CrossRef]
- Zhang, J.; Rodríguez, F.; Navas, M.J.; Costa-Hurtado, M.; Almagro, V.; Bosch-Camós, L.; López, E.; Cuadrado, R.; Accensi, F.; Pina-Pedrero, S.; et al. Fecal microbiota transplantation from warthog to pig confirms the influence of the gut microbiota on African swine fever susceptibility. Sci. Rep. 2020, 10, 17605. [Google Scholar] [CrossRef]
- Seib, K.L.; Zhao, X.; Rappuoli, R. Developing vaccines in the era of genomics: A decade of reverse vaccinology. Clin. Microbiol. Infect. 2012, 18, 109–116. [Google Scholar] [CrossRef]
- Bragazzi, N.L.; Gianfredi, V.; Villarini, M.; Rosselli, R.; Nasr, A.; Hussein, A.; Martini, M.; Behzadifar, M. Vaccines Meet Big Data: State-of-the-Art and Future Prospects. From the Classical 3Is (“Isolate–Inactivate–Inject”) Vaccinology 1.0 to Vaccinology 3.0, Vaccinomics, and Beyond: A Historical Overview. Front. Public Health 2018, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, N.C.; Maman, Y.; Kohlbacher, O.; Louzoun, Y. Universal peptide vaccines—Optimal peptide vaccine design based on viral sequence conservation. Vaccine 2011, 29, 8745–8753. [Google Scholar] [CrossRef] [PubMed]
- Argilaguet, J.M.; Pérez-Martín, E.; Gallardo, C.; Salguero, F.J.; Borrego, B.; Lacasta, A.; Accensi, F.; Díaz, I.; Nofrarías, M.; Pujols, J.; et al. Enhancing DNA immunization by targeting ASFV antigens to SLA-II bearing cells. Vaccine 2011, 29, 5379–5385. [Google Scholar] [CrossRef] [PubMed]
- Sunwoo, S.-Y.; Pérez-Núñez, D.; Morozov, I.; Sánchez, E.G.; Gaudreault, N.N.; Trujillo, J.D.; Mur, L.; Nogal, M.; Madden, D.; Urbaniak, K.; et al. DNA-Protein Vaccination Strategy Does Not Protect from Challenge with African Swine Fever Virus Armenia 2007 Strain. Vaccines 2019, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Masignani, V.; Pizza, M.; Moxon, E.R. The development of a vaccine against Meningococcus B using reverse vaccinology. Front. Immunol. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 623–656. [Google Scholar] [CrossRef]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Liu, Q.; Ma, B.; Qian, N.; Zhang, F.; Tan, X.; Lei, J.; Xiang, Y. Structure of the African swine fever virus major capsid protein p72. Cell Res. 2019, 29, 953–955. [Google Scholar] [CrossRef]
- Dhanda, S.K.; Mahajan, S.; Paul, S.; Yan, Z.; Kim, H.; Jespersen, M.C.; Jurtz, V.; Andreatta, M.; Greenbaum, J.A.; Marcatili, P.; et al. IEDB-AR: Immune epitope database—Analysis resource in 2019. Nucleic Acids Res. 2019, 47, W502–W506. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.A.; Guan, P.; Flower, D.R. EpiJen: A server for multistep T cell epitope prediction. BMC Bioinform. 2006, 7, 131. [Google Scholar] [CrossRef] [PubMed]
- Peters, B.; Bulik, S.; Tampe, R.; van Endert, P.M.; Holzhütter, H.-G. Identifying MHC Class I Epitopes by Predicting the TAP Transport Efficiency of Epitope Precursors. J. Immunol. 2003, 171, 1741–1749. [Google Scholar] [CrossRef] [PubMed]
- Moutaftsi, M.; Peters, B.; Pasquetto, V.; Tscharke, D.C.; Sidney, J.; Bui, H.-H.; Grey, H.; Sette, A. A consensus epitope prediction approach identifies the breadth of murine TCD8+-cell responses to vaccinia virus. Nat. Biotechnol. 2006, 24, 817–819. [Google Scholar] [CrossRef] [PubMed]
- Kotturi, M.F.; Peters, B.; Buendia-Laysa, F.; Sidney, J.; Oseroff, C.; Botten, J.; Grey, H.; Buchmeier, M.J.; Sette, A. The CD8+ T-Cell Response to Lymphocytic Choriomeningitis Virus Involves the L Antigen: Uncovering New Tricks for an Old Virus. J. Virol. 2007, 81, 4928–4940. [Google Scholar] [CrossRef] [PubMed]
- OIE African Swine Fever (ASF) Report N° 56: October 16 to October 29, 2020. Available online: https://www.oie.int/fileadmin/Home/eng/Animal_Health_in_the_World/docs/pdf/Disease_cards/ASF/Report_56_Current_situation_of_ASF.pdf (accessed on 9 November 2020).
- FAO ASF Situation Update. Available online: http://www.fao.org/ag/againfo/programmes/en/empres/ASF/situation_update.html (accessed on 9 November 2020).
- Rabobank Rising African Swine Fever Losses to Lift All Protein Boats. Available online: https://research.rabobank.com/far/en/sectors/animal-protein/rising-african-swine-fever-losses-to-lift-all-protein.html (accessed on 12 December 2020).
- Reuters Special Report: Before Coronavirus, China Bungled Swine Epidemic with Secrecy. Available online: https://www.reuters.com/article/us-swinefever-china-epidemic-specialrepo-idUSKBN20S189 (accessed on 9 November 2020).
- Leitão, A.; Cartaxeiro, C.; Coelho, R.; Cruz, B.; Parkhouse, R.M.E.; Portugal, F.C.; Vigário, J.D.; Martins, C.L.V. The non-haemadsorbing African swine fever virus isolate ASFV/NH/P68 provides A model for defining the protective anti-virus immune response. J. Gen. Virol. 2001, 82, 513–523. [Google Scholar] [CrossRef]
- Borca, M.V.; O’Donnell, V.; Holinka, L.G.; Risatti, G.R.; Ramirez-Medina, E.; Vuono, E.A.; Shi, J.; Pruitt, S.; Rai, A.; Silva, E.; et al. Deletion of CD2-like gene from the genome of African swine fever virus strain Georgia does not attenuate virulence in swine. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef]
- Moyle, P.M.; Toth, I. Modern Subunit Vaccines: Development, Components, and Research Opportunities. ChemMedChem 2013, 8, 360–376. [Google Scholar] [CrossRef]
- Gaudreault, N.N.; Richt, J.A. Subunit vaccine approaches for African swine fever virus. Vaccines 2019, 7, 56. [Google Scholar] [CrossRef]
- Sheikh, Q.M.; Gatherer, D.; Reche, P.A.; Flower, D.R. Towards the knowledge-based design of universal influenza epitope ensemble vaccines. Bioinformatics 2016, 32, 3233–3239. [Google Scholar] [CrossRef]
- Escribano, J.M.; Galindo, I.; Alonso, C. Antibody-mediated neutralization of African swine fever virus: Myths and facts. Virus Res. 2013, 173, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Quinzo, M.J.; Lafuente, E.M.; Zuluaga, P.; Flower, D.R.; Reche, P.A. Computational assembly of a human Cytomegalovirus vaccine upon experimental epitope legacy. BMC Bioinform. 2019, 20, 476. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Padilla, J.; Lafuente, E.M.; Reche, P.A. Computer-Aided Design of an Epitope-Based Vaccine against Epstein-Barr Virus. J. Immunol. Res. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Bresciani, A.; Greenbaum, J.; Arlehamn, C.S.L.; Sette, A.; Peters, B.; Nielsen, M. The interplay of sequence conservation and T cell immune recognition. In Proceedings of the 5th ACM Conference on Bioinformatics, Computational Biology, and Health Informatics, BCB’, Newport Beach, CA, USA, 20–22 September 2014; pp. 739–743. [Google Scholar] [CrossRef]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef]
- Rodríguez, J.M.; Yáñez, R.J.; Almazán, F.; Viñuela, E.; Rodriguez, J.F. African swine fever virus encodes a CD2 homolog responsible for the adhesion of erythrocytes to infected cells. J. Virol. 1993, 67, 5312–5320. [Google Scholar] [CrossRef]
- Chapman, D.A.G.; Tcherepanov, V.; Upton, C.; Dixon, L.K. Comparison of the genome sequences of non-pathogenic and pathogenic African swine fever virus isolates. J. Gen. Virol. 2008, 89, 397–408. [Google Scholar] [CrossRef]
- Andrés, G.; García-Escudero, R.; Viñuela, E.; Salas, M.L.; Rodríguez, J.M. African Swine Fever Virus Structural Protein pE120R Is Essential for Virus Transport from Assembly Sites to Plasma Membrane but Not for Infectivity. J. Virol. 2001, 75, 6758–6768. [Google Scholar] [CrossRef]
- Salas, M.L.; Andrés, G. African swine fever virus morphogenesis. Virus Res. 2013, 173, 29–41. [Google Scholar] [CrossRef]
- Gómez-Puertas, P.; Rodríguez, F.; Oviedo, J.M.; Ramiro-Ibáñez, F.; Ruiz-Gonzalvo, F.; Alonso, C.; Escribano, J.M. Neutralizing antibodies to different proteins of African swine fever virus inhibit both virus attachment and internalization. J. Virol. 1996, 70, 5689–5694. [Google Scholar] [CrossRef]
- Hühr, J.; Schäfer, A.; Schwaiger, T.; Zani, L.; Sehl, J.; Mettenleiter, T.C.; Blome, S.; Blohm, U. Impaired T cell responses in domestic pigs and wild boar upon infection with a highly virulent African swine fever virus strain. Transbound. Emerg. Dis. 2020, 67, 3016–3032. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J.; Lee, C.Y.; Ibrahim, S.; Watts, P.; Shlomchik, M.; Weigert, M.; Litwin, S. A Shannon entropy analysis of immunoglobulin and T cell receptor. Mol. Immunol. 1997, 34, 1067–1082. [Google Scholar] [CrossRef]
- Michel-Todó, L.; Bigey, P.; Reche, P.A.; Pinazo, M.-J.; Gascón, J.; Alonso-Padilla, J. Design of an epitope-based vaccine ensemble for animal trypanosomiasis by computational methods. Vaccines 2020, 8, 130. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Lundegaard, C.; Lund, O.; Keşmir, C. The role of the proteasome in generating cytotoxic T-cell epitopes: Insights obtained from improved predictions of proteasomal cleavage. Immunogenetics 2005, 57, 33–41. [Google Scholar] [CrossRef]
- Besser, H.; Louzoun, Y. Cross-modality deep learning-based prediction of TAP binding and naturally processed peptide. Immunogenetics 2018, 70, 419–428. [Google Scholar] [CrossRef]
- Greenbaum, J.; Sidney, J.; Chung, J.; Brander, C.; Peters, B.; Sette, A. Functional classification of class II human leukocyte antigen (HLA) molecules reveals seven different supertypes and a surprising degree of repertoire sharing across supertypes. Immunogenetics 2011, 63, 325–335. [Google Scholar] [CrossRef]
- Farrell, D.; Jones, G.; Pirson, C.; Malone, K.; Rue-Albrecht, K.; Chubb, A.J.; Vordermeier, M.; Gordon, S.V. Integrated computational prediction and experimental validation identifies promiscuous T cell epitopes in the proteome of Mycobacterium bovis. Microb. Genom. 2016, 2, e000071. [Google Scholar] [CrossRef]
- Emanuelsson, O.; Brunak, S.; von Heijne, G.; Nielsen, H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2007, 2, 953–971. [Google Scholar] [CrossRef]
- Almagro Armenteros, J.J.; Sønderby, C.K.; Sønderby, S.K.; Nielsen, H.; Winther, O. DeepLoc: Prediction of protein subcellular localization using deep learning. Bioinformatics 2017, 33, 3387–3395. [Google Scholar] [CrossRef]
- Pierleoni, A.; Martelli, P.L.; Casadio, R. PredGPI: A GPI-anchor predictor. BMC Bioinform. 2008, 9, 392. [Google Scholar] [CrossRef]
- Hubbard, S.J.; Thornton, J.M. “NACCESS” Computer Program; Department of Biochemistry and Molecular Biology; University College London: London, UK, 1993. [Google Scholar]
- The Python Software Foundation Python. Available online: https://www.python.org/ (accessed on 9 November 2020).
- National Center for Biotechnology Information National Center for Biotechnology Information (NCBI). Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 15 June 2020).
- Camacho, C.; Madden, T.; Tao, T.; Agarwala, R.; Morgulis, A. BLAST Command Line Applications User Manual; National Center for Biotechnology Information: Bethesda, MD, USA, 2019.
- Correa-Fiz, F.; Fraile, L.; Aragon, V. Piglet nasal microbiota at weaning may influence the development of Glässer’s disease during the rearing period. BMC Genom. 2016, 17, 404. [Google Scholar] [CrossRef] [PubMed]
- Correa-Fiz, F.; Blanco-Fuertes, M.; Navas, M.J.; Lacasta, A.; Bishop, R.P.; Githaka, N.; Onzere, C.; Le Potier, M.-F.; Almagro-Delgado, V.; Martinez, J.; et al. Comparative analysis of the fecal microbiota from different species of domesticated and wild suids. Sci. Rep. 2019, 9, 13616. [Google Scholar] [CrossRef] [PubMed]
- Mahmmod, Y.S.; Correa-Fiz, F.; Aragon, V. Variations in association of nasal microbiota with virulent and non-virulent strains of Glaesserella (Haemophilus) parasuis in weaning piglets. Vet. Res. 2020, 51, 7. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Epitope | Protein Name (ORF) | Protein ID 1 | Sus scrofa Hit (id %) 2 | Microbiota Hit (id %) 2 |
|---|---|---|---|---|
| FPENSHNIQTAGKQD | p72 (B646L) | E0WMM0 | XP_020938976.1 (40.0%) | WP_008983921.1 (53.3%) |
| YCEYPGERLYENVRFDVNGNSLDEYSSDVTTL | p72 (B646L) | E0WMM0 | XP_020953642.1 (25.0%) | WP_091148772.1 (34.4%) |
| VCKVDKDCGSGEHCV | p22 (KP177R) | E0WMB7 | XP_020923036.1 (66.7%) | WP_046972048.1 (60.0%) |
| AAIEEEDIQFINPYQD | p54 (E183L) | E0WM75 | XP_020932305.1 (50.0%) | WP_042346777.1 (68.8%) |
| Epitope | Protein Name (ORF) | Protein ID 1 | Sus scrofa Hit (id %) 2 | Microbiota Hit (id %) 2 |
|---|---|---|---|---|
| LATCGKAGNFCECSNYSTS | CD2 homolog (E402R) | E0WMJ6 | XP_003483701.1 (47.4%) | WP_007633917.1 (47.4%) |
| KKQQPPKKVCKVDKDCGSGEHC | p22 (KP177R) | E0WMB7 | XP_020923055.1 (40.9%) | WP_137027291.1 (63.6%) |
| FFQPVYPRHYGECLSP | p54 (E183L) | E0WM75 | XP_020934554.1 (50.0%) | WP_018576392.1 (56.3%) |
| GFEYNKVRPHTGTPTLGNKLT | p72 (B646L) | E0WMM0 | XP_020939243.1 (42.9%) | WP_143542922.1 (47.6%) |
| HKPHQSKPILTDENDTQRTC | p72 (B646L) | E0WMM0 | XP_020932317.1 (35.0%) | WP_169883328.1 (45.0%) |
| HTNPKFLSQHFPENSHNIQTAGKQDITPITD | p72 (B646L) | E0WMM0 | XP_020933726.1 (32.3%) | WP_042032899.1 (38.7%) |
| QMGAHGQLQTFPRNGYDWDNQTPLE | p72 (B646L) | E0WMM0 | XP_020956625.1 (28.0%) | WP_075838746.1 (36.0%) |
| SFQDRDTALPDACSSISDI | p72 (B646L) | E0WMM0 | XP_020950058.1 (42.1%) | WP_165776787.1 (57.9%) |
| TWNISDQNPHQHRDWHK | p72 (B646L) | E0WMM0 | XP_003123199.2 (41.2%) | WP_077451423.1 (58.8%) |
| VTHTNNNHHDEKLMS | p72 (B646L) | E0WMM0 | XP_020946598.1 (46.7%) | WP_072834091.1 (53.3%) |
| TSPLLSHNLSTREGIKQ | p17 (D117L) | E0WM57 | XP_020947378.1 (47.1%) | WP_021704232.1 (58.8%) |
| GVCKNKVFEKHPLIKKNDY | MGF_110-9L | E0WMD4 | XP_020922529.1 (47.4%) | WP_124535981.1 (63.2%) |
| AGRGIPLGNPHVKPNIEQELIKS | p49 (B438L) | E0WML4 | XP_013850764.1 (30.4%) | WP_168007327.1 (56.5%) |
| FPKDFNASSVPLTSAEKDHSLRGDNS | p49 (B438L) | E0WML4 | NP_999435.1 (50.0%) | WP_007759066.1 (46.2%) |
| GQAEYFDTSKQTISRHNNYIPKYTGGIGDS | p49 (B438L) | E0WML4 | XP_020939832.1 (33.3%) | WP_095912160.1 (36.7%) |
| LADYRSDPPLWESDLPRHNRYSDNILN | p49 (B438L) | E0WML4 | XP_020954566.1 (25.9%) | WP_143667676.1 (55.6%) |
| LNPQHKNIGYGDAQDLEPYS | p49 (B438L) | E0WML4 | XP_013849660.1 (30.0%) | WP_085490987.1 (60.0%) |
| PSFDNDVKRRNKDTVWARFGV | Trans-prenyltransferase (B318L) | E0WML3 | XP_005674076.1 (38.1%) | WP_142159516.1 (47.6%) |
| GINNLGEKIYTCEPFKTSF | Transmembrane protein B169L | E0WML5 | XP_003354140.1 (42.1%) | WP_095898538.1 (63.2%) |
| KYLERQDLELLGYSPT | Transmembrane protein C257L | E0WMK4 | XP_003356508.3 (68.8%) | WP_111879816.1 (62.5%) |
| DEPIVQNPFLENFWKPEQKTFNQSGLFEESS | Uncharacterized protein B475L | E0WML6 | XP_020932571.1 (29.0%) | WP_078933170.1 (38.7%) |
| FRDAQNPPSSFTLGGHCQA | Uncharacterized protein E146L | E0WM78 | NP_001121958.1 (52.6%) | WP_147756634.1 (47.4%) |
| EASYDTMRTKLMKFSGINKEKENN | Uncharacterized protein M1249L | E0WMJ8 | XP_020928637.1 (41.7%) | WP_022932625.1 (41.7%) |
| GKQEAELITTEYLNIKKQWELQEKNACA | Uncharacterized protein M1249L | E0WMJ8 | XP_020951797.1 (50.0%) | WP_134792973.1 (46.4%) |
| ISAYSSPGLFGEDIID | Uncharacterized protein M1249L | E0WMJ8 | XP_020934753.1 (50.0%) | WP_041516260.1 (56.3%) |
| LSQPELVHDYYNNYKDQY | Uncharacterized protein M1249L | E0WMJ8 | XP_020923164.1 (38.9%) | WP_133014505.1 (55.6%) |
| NPVEQKFLQHAEQREKEQMILQ | Uncharacterized protein M1249L | E0WMJ8 | XP_020946579.1 (50.0%) | WP_085491731.1 (45.5%) |
| Epitope | Protein (ORF) | Protein ID 1 | Mean Flex. 2 | Mean RSA 3 | Sus scrofa Hit (id %) 4 | Microbiota Hit (id %) 4 |
|---|---|---|---|---|---|---|
| KPHQSKPILTDENDTQRT | p72 (B646L) | E0WMM0 | 1.7 | 50.1 | XP_020932317.1 (38.9%) | WP_169883328.1 (50.0%) |
| NIQTAGKQDITPITD | p72 (B646L) | E0WMM0 | 1.8 | 56.8 | XP_020933726.1 (66.7%) | WP_001886209.1 (53.3%) |
| Epitope | No. HLA Alleles 1 | Protein Name (ORF) | Protein ID 2 | Sus scrofa Hit (id %) 3 | Microbiota Hit (id %) 3 |
|---|---|---|---|---|---|
| GLGFILIVIFIYLLLITLQQMLTRHI | 5 | Uncharacterized protein (B117L) | E0WM34 | XP_003360623.1 (46.2%) | WP_119317083.1 (53.9%) |
| MNIYLVWFLYILLGNLILAVIY | 4 | ASFV_G_ACD_01990 | E0WMB3 | XP_003124218.1 (59.1%) | WP_155703379.1 (68.2%) |
| LSLICVFSHFFEELYITKP | 3 | Probable methyltransferase (EP424R) | E0WMJ3 | XP_013843985.2 (42.1%) | WP_010770249.1 (52.6%) |
| WYLKYVIAYILLLTMLVIGLIYRIIVLIYRSIQAQK | 3 | ASFV_G_ACD_01940 | E0WMA9 | XP_013836670.2 (33.3%) | WP_100215420.1 (55.6%) |
| AINFLLLQNGSAVLRYS | 2 | p72 (B646L) | E0WMM0 | NP_999474.1 (52.9%) | WP_113638931.1 (64.7%) |
| ATRLVAVRAQQLAINGSTMLKKK | 2 | DNA-directed RNA polymerase subunit 6 homolog (C147L) | E0WMK7 | XP_020956260.1 (30.4%) | WP_093129025.1 (56.5%) |
| GLNFQAVRYEMIMSLPLDIP | 2 | Putative ATP-dependent RNA helicase (D1133L) | E0WM56 | XP_013834167.2 (55.0%) | WP_096721649.1 (60%) |
| HYPASFHYTMLEALIIDN | 2 | Putative ATP-dependent RNA helicase (D1133L) | E0WM56 | XP_005666131.2 (38.9%) | WP_167872395.1 (55.6%) |
| ISIITFLSLRKRKKHVEEI | 2 | CD2 homolog (E402R) | E0WMJ6 | XP_020951930.1 (42.1%) | WP_039931062.1 (52.6%) |
| MYFQQTRSILIKNDAVFILNLG | 2 | ASFV_G_ACD_01870 | E0WMA2 | XP_020949404.1 (54.6%) | WP_069997539.1 (40.9%) |
| NAFVDYIISNFNHAVTCRKP | 2 | Transmembrane protein (B66L) | E0WM38 | XP_020936233.1 (40.0%) | WP_128359657.1 (45.0%) |
| NNILVEILSFKNYYSSNTSLLSIKT | 2 | MGF_360-21R | E0WMB1 | XP_020924396.1 (40.0%) | WP_117888406.1 (44.0%) |
| NVIFLKVISNTAVSVFWRD | 2 | Uncharacterized protein (C62L) | E0WMK8 | XP_020958320.1 (52.6%) | WP_094566288.1 (52.6%) |
| PTPLIPSMAMSIPRMINKRKKRIQFLTFLTNLFLYN | 2 | Uncharacterized protein (A118R) | E0WMH4 | XP_020945397.1 (30.6%) | WP_165296256.1 (50.0%) |
| Predicted Epitope | No. SLA Alleles 1 | Protein (ORF) | Protein ID 2 | TAP IC50 3 | Sus scrofa Hit (id %) 4 | Microbiota Hit (id %)4 |
|---|---|---|---|---|---|---|
| MAMQKLFTY | 29 | Putative DNA-directed RNA polymerase subunit 5 homolog (D205R) | E0WM58 | −0.94 | NP_001037992.1 (66.7%) | WP_025027576.1 (77.8%) |
| KRHENIWML | 27 | Uncharacterized protein D339L | E0WM55 | −2.81 | XP_013850843.2 (55.6%) | WP_130548297.1 (77.8%) |
| CTQPARVTY | 22 | DNA-directed RNA polymerase subunit beta (EP1242L) | E0WMJ1 | 0.17 | XP_003127876.2 (66.7%) | WP_164721234.1 (77.8%) |
| NIMPGLVSY | 21 | Ribonucleoside-diphosphate reductase (F334L) | E0WMI2 | 0.65 | XP_020927225.1 (55.6%) | WP_157084930.1 (77.8%) |
| ANPSEGWKY | 10 | DNA topoisomerase 2 (P1192R) | E0WM62 | 0.23 | XP_020958373.1 (55.6%) | WP_117029589.1 (77.8%) |
| EEFNYLWVY | 7 | Uncharacterized protein G1340L | E0WM39 | −2.05 | NP_999483.1 (66.7%) | WP_083089560.1 (77.8%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ros-Lucas, A.; Correa-Fiz, F.; Bosch-Camós, L.; Rodriguez, F.; Alonso-Padilla, J. Computational Analysis of African Swine Fever Virus Protein Space for the Design of an Epitope-Based Vaccine Ensemble. Pathogens 2020, 9, 1078. https://doi.org/10.3390/pathogens9121078
Ros-Lucas A, Correa-Fiz F, Bosch-Camós L, Rodriguez F, Alonso-Padilla J. Computational Analysis of African Swine Fever Virus Protein Space for the Design of an Epitope-Based Vaccine Ensemble. Pathogens. 2020; 9(12):1078. https://doi.org/10.3390/pathogens9121078
Chicago/Turabian StyleRos-Lucas, Albert, Florencia Correa-Fiz, Laia Bosch-Camós, Fernando Rodriguez, and Julio Alonso-Padilla. 2020. "Computational Analysis of African Swine Fever Virus Protein Space for the Design of an Epitope-Based Vaccine Ensemble" Pathogens 9, no. 12: 1078. https://doi.org/10.3390/pathogens9121078
APA StyleRos-Lucas, A., Correa-Fiz, F., Bosch-Camós, L., Rodriguez, F., & Alonso-Padilla, J. (2020). Computational Analysis of African Swine Fever Virus Protein Space for the Design of an Epitope-Based Vaccine Ensemble. Pathogens, 9(12), 1078. https://doi.org/10.3390/pathogens9121078