1. Introduction
Piscine orthoreovirus 1 (PRV-1) is the causative agent of heart and skeletal muscle inflammation (HSMI), an important disease in farmed Atlantic salmon (
Salmo salar) [
1,
2,
3]. However, all PRV-1 infected fish do not develop HSMI. This indicates that unknown viral, host or environmental factors are important for disease development.
In 1999, the first outbreak of HSMI was described in farmed Atlantic salmon in Norway [
1]. Subsequently, the disease spread along the coast and is now causing numerous outbreaks each year in Norway [
4]. The disease is characterized mainly by inflammation in the heart and skeletal muscle, with outbreaks of high morbidity and mortality up to 20% [
2]. PRV-1 was identified in diseased fish [
3] and later shown to be the causative agent of HSMI [
2]. Interestingly, the virus has been revived from Norwegian archival samples dating back to 1988 [
5], 11 years prior to the appearance of the disease. This has led to a hypothesis that virus evolution has increased the virulence of PRV-1, which in turn has contributed to the emergence of HSMI in Norway [
5].
PRV-1 is almost ubiquitous in countries with large scale farming of Atlantic salmon but HSMI does not appear to be. Westcoast Canada is a notable example; the virus is readily detected in farms but only a few cases of HSMI have been reported [
6]. Similarly, the Faroe Islands have reported the virus but not the disease [
5]. This contrasts the high prevalence of HSMI observed in Norway [
4], as well as observations of HSMI in Scotland and Chile [
7,
8].
Experimental trials performed with Norwegian and Canadian PRV-1 isolates have given different results. HSMI has been reproduced with Norwegian isolates in a number of experimental trials [
2,
9,
10,
11], whereas similar experiments with Canadian isolates have not [
12,
13]. All trials describe an early acute phase characterized by high viral load in blood [
2,
12,
13], when PRV-1 replicates in its major target cell erythrocytes [
14]. The major difference is the subsequent development of heart lesions observed with Norwegian isolates [
2,
9], which is almost absent with Canadian isolates [
12,
13]. Furthermore, challenge experiments with Canadian isolates have reported no virus in plasma and only minor antiviral immune responses during replication in blood [
12,
13]. Similar experiments with Norwegian isolates have shown high plasma viremia and a robust innate immune response in blood cells [
2,
15]. These observations have led to a hypothesis that there are virulence differences between Norwegian and Canadian PRV-1 isolates. However, experimental testing of the hypothesis requires that the isolates are compared in a trial where host and environmental factors as well as viral inoculum are all standardized.
Differences in virulence are reflected in the nucleotide sequence of the viral genome. PRV-1 belongs to the genus
Orthoreovirus in the family Reoviridae [
16,
17] and has a ten-segmented double-stranded RNA (dsRNA) genome packed in a double protein capsid [
18]. The three large (L1–3) three medium (M1–3) and four small (S1–4) segments each encodes one protein named with its corresponding Greek letter λ, μ and σ, except segment S1, which is biscistronic [
16]. Through point mutations, reassortment and recombination these viruses will naturally explore the available sequence space and natural selection will continuously optimize their fitness to changing intra- and extracellular conditions [
19]. Phenotypical properties, such as induction of HSMI, can be linked to single or multiple mutations or reassortment involving one or several segments. PRV-1 sequences that are available in accessible depositaries mostly include only the S1 sequence and without a description of the disease history of the fish population from where the virus isolate originated, which together limits the utility of these nucleotide sequences in virulence investigations. The number of whole genome PRV-1 sequences are increasing and in silico studies have identified segment S1 and M2 as important for the development of HSMI [
5]. Furthermore, comparison of Norwegian and Pacific Canadian isolates used in challenge trials have shown that most sequence variation is found in segment S1 [
12].
The aim of this study was to determine virulence differences in Atlantic salmon between PRV-1 isolates. Due to PRV-1′s resistance to be propagated in cell cultures, recent methodologic advancement for virus purification was utilized to standardize the challenge dose in a trial comparing two Norwegian field isolates from 2018, three historical Norwegian isolates predating discovery of HSMI and one Canadian isolate. The results demonstrated virulence differences between PRV-1 isolates regarding their ability to induce HSMI.
3. Discussion
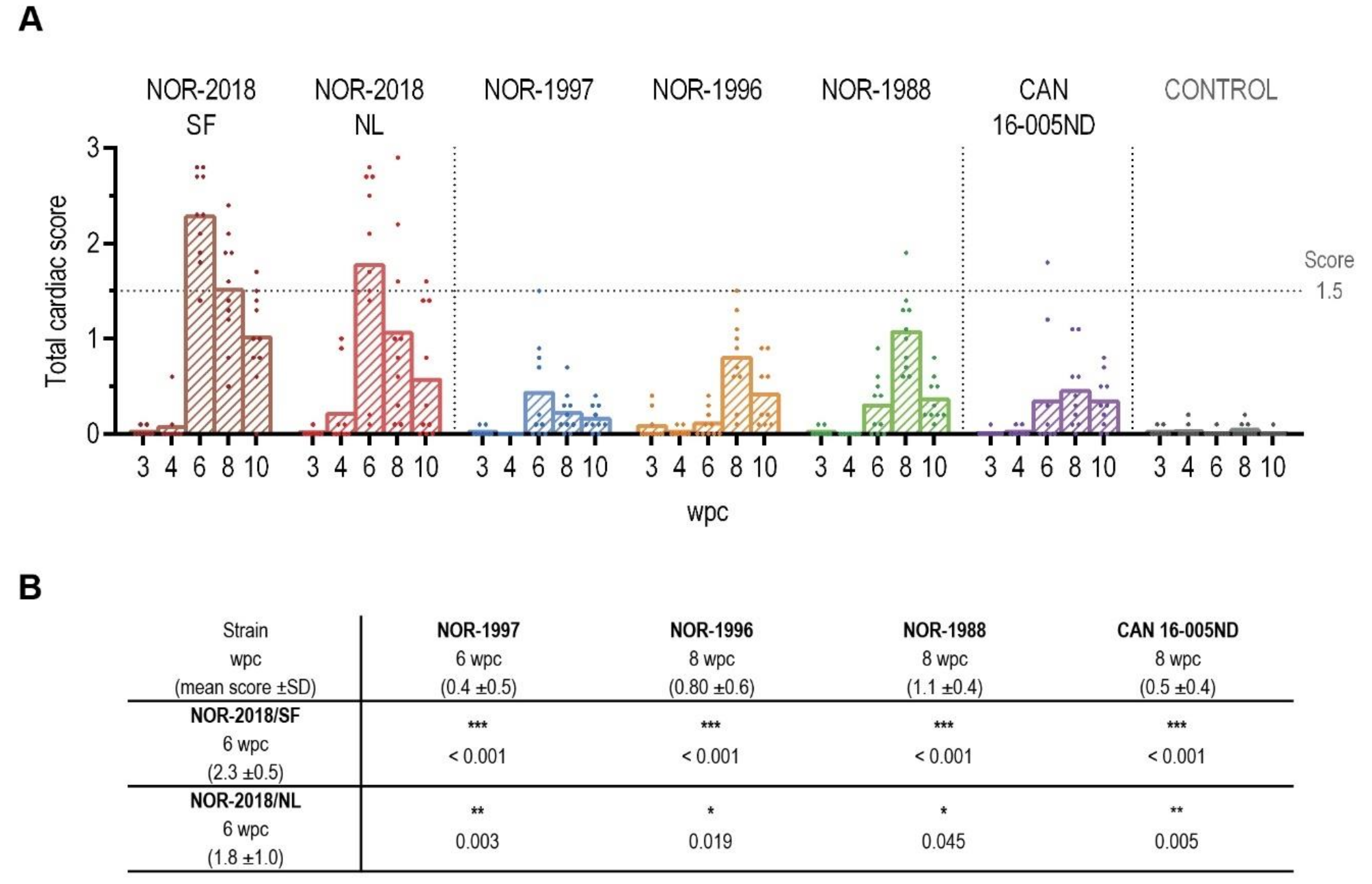
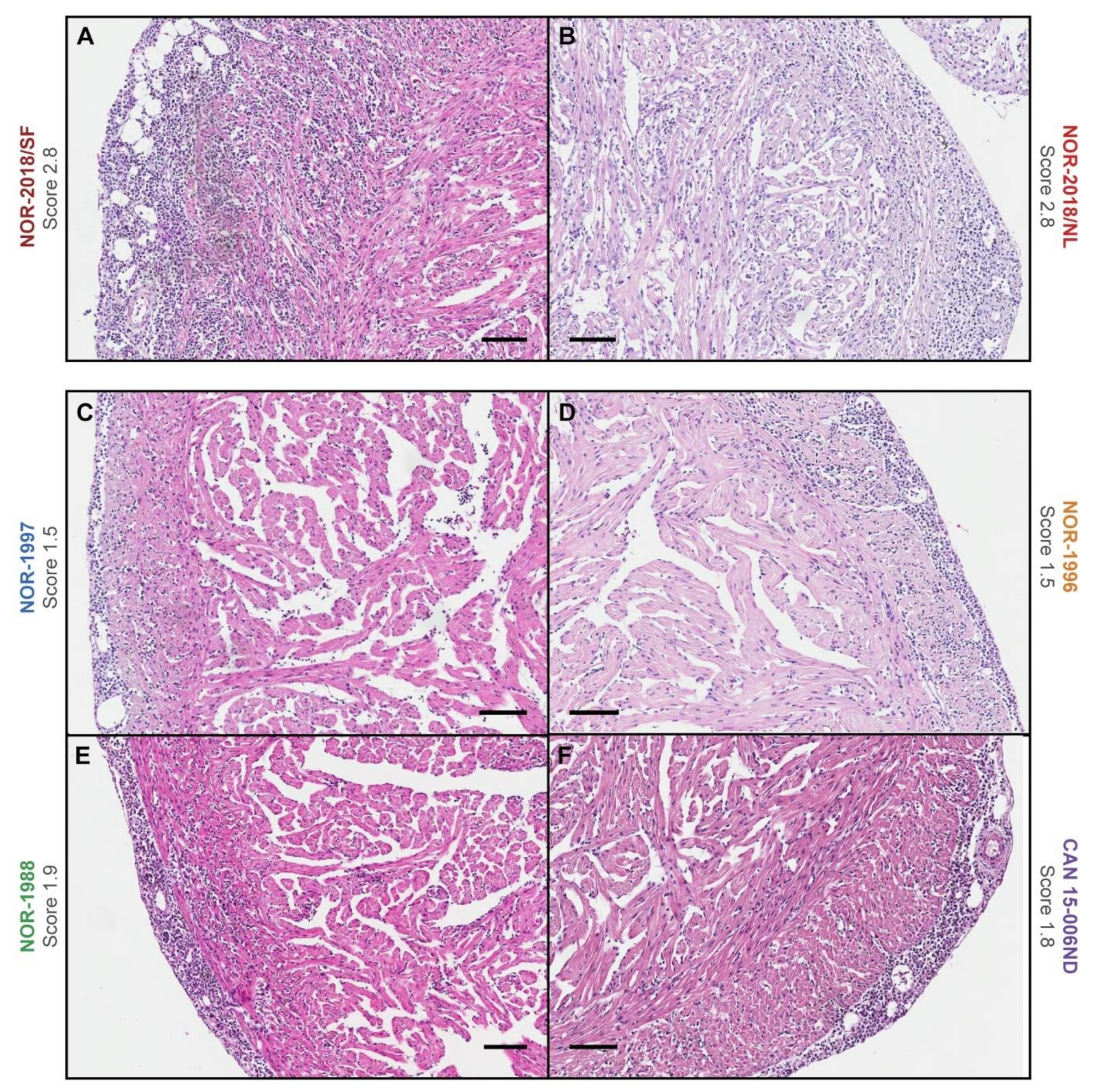
In this study a dose standardized challenge trial demonstrated that there are virulence differences between PRV-1 isolates in Atlantic salmon regarding induction of histopathological changes typical for HSMI. The high virulent isolates, that is, two Norwegian field isolates (NOR-2018/SF, NOR-2018/NL), induced cardiac lesions consistent with HSMI. The low virulent isolates, that is, three historical Norwegian isolates (NOR-1997, NOR-1996, NOR-1988) and one Canadian isolate (CAN 16-005ND) induced mainly mild cardiac lesions.
Conflicting results have been reported in the field of PRV-1 research with both success [
2,
21] and failure [
12,
13] to produce cardiac lesions consistent with HSMI. Our results suggest that this dichotomy can be at least partly explained by virulence differences. The severe cardiac lesions induced by the Norwegian 2018 field isolates in the present study are consistent with those observed in previous trials using another contemporary Norwegian field isolate (NOR-2012) [
2]. In contrast, only mild lesions were induced by the CAN 16-005ND isolate, which is comparable to the lesions observed in a previous trial using two Canadian isolates, including the CAN 16-005ND isolate [
12]. Similar low-grade lesions were observed for the historical Norwegian isolates that had been collected prior the emergence of HSMI, which support the hypothesis that PRV-1 isolates present in Norway before HSMI was first recognized in 1999 were of low virulence. However, our results suggest that all PRV-1 isolates can induce cardiac lesions but of different severity.
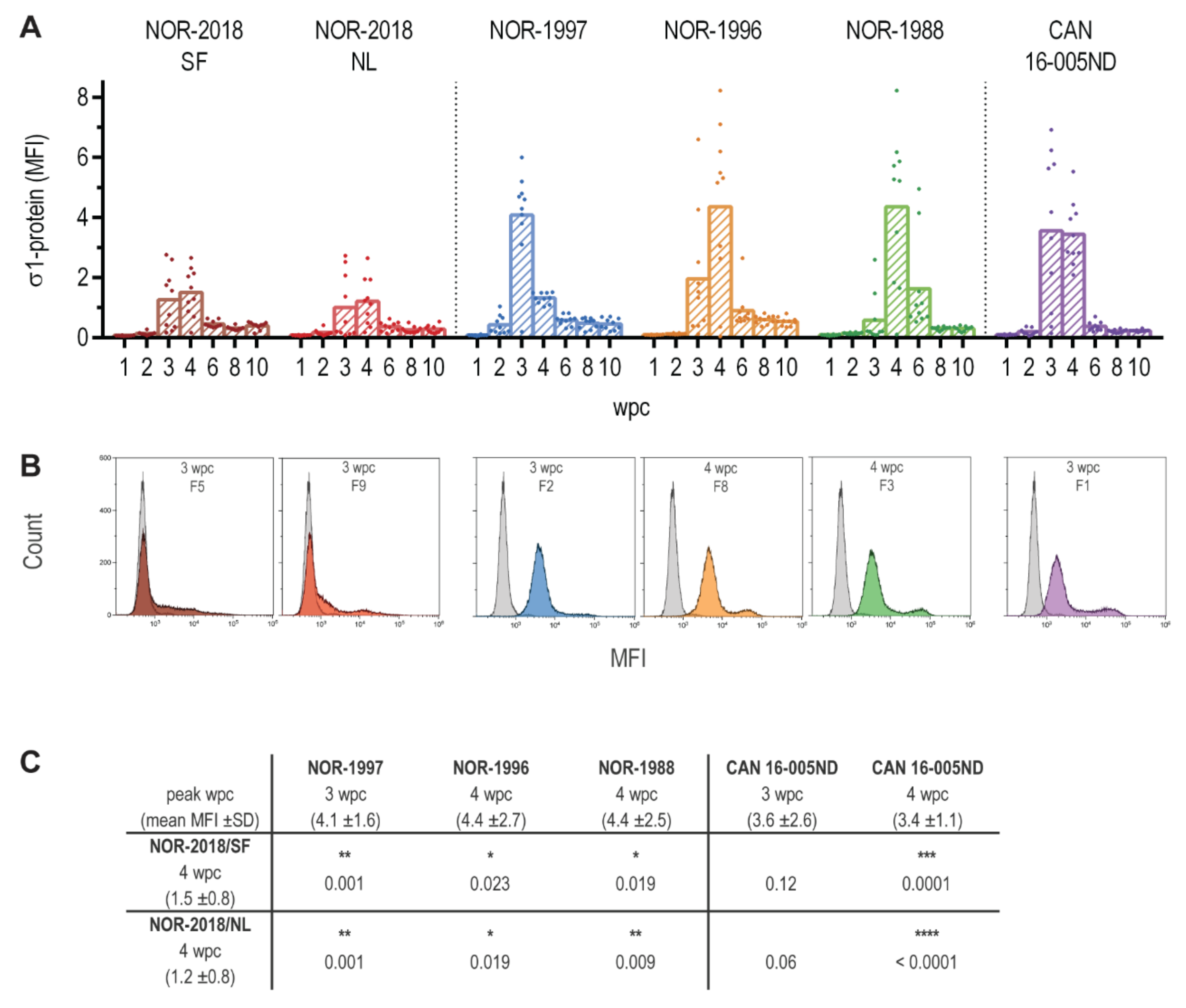
High loads of PRV-1 in blood cells have previously been well documented after infection with both Norwegian and Canadian isolates [
2,
12,
13]. The present study confirmed this general finding but also demonstrated differences between the virus isolates. For the low virulent isolates, all blood cells were PRV positive with a higher total load of viral protein in blood cells compared to the high virulent isolates in which virus could be detected in only a subpopulation of blood cells. These differences could not be seen by the detection of viral RNA in blood cells. However, the protein detection did not differentiate between intracellular or surface bound virus and targeted a structural protein. Mammalian reoviruses are known to bind sialic acid which is abundant on the erythrocyte surface [
22,
23]. Combining surface and intracellular staining of blood cells including detection of both structural and non-structural viral proteins, could possibly identify the cell population with replicating virus.
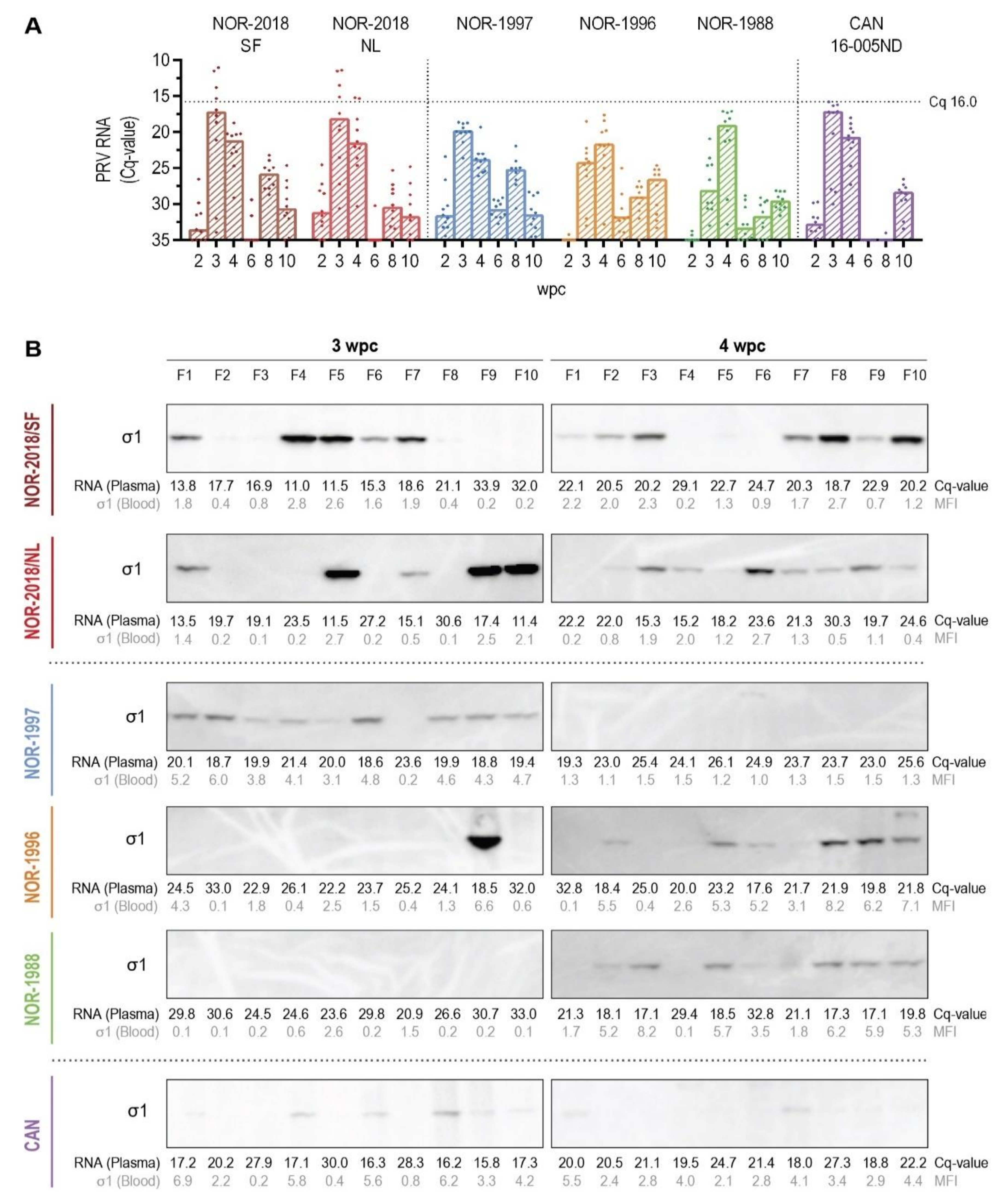
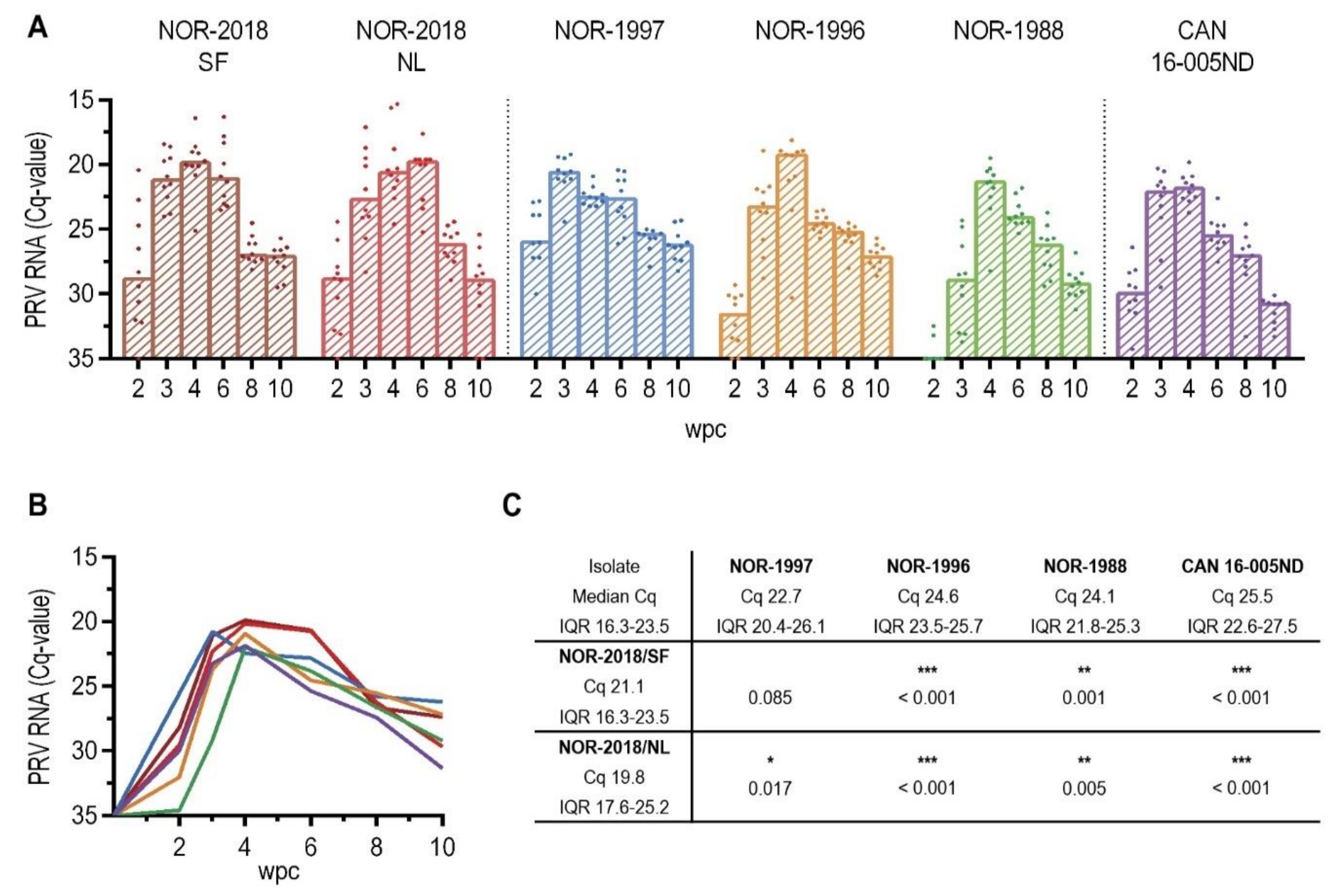
Interestingly, a higher plasma viremia was detected for the high virulent isolates (NOR-2018 isolates) than for the low virulent isolates (historical Norwegian and Canadian isolates). Previous studies of Canadian isolates have not detected PRV-1 in plasma during infection [
12] but have been reported using Norwegian isolates [
2,
11,
14]. In our study, virus was detected in plasma at peak of infection in all isolates, including all low virulent isolates but the latter with lower amount as confirmed by detection of viral RNA and multiple viral proteins. The amount of plasma viremia is an important factor with a potential direct effect on pathogenesis and shedding and could be a defining virulence factor. The difference in plasma viremia between high and low virulent isolates could either indicate a more efficient release of high virulent isolates from infected cells or reflect more efficient attachment of low virulent isolates to the erythrocytic surface. Release of virus could be caused by lytic or non-lytic shedding from infected cells or possibly through the process of removal of infected erythrocytes from the circulation, in the spleen. The high amount of virus in plasma following infection with high virulent isolates did not coincide with reduced hemoglobin concentration. This could indicate a non-lytic exit of virus. Although non-enveloped viruses usually cause lysis of the infected cell, non-lytic shedding has recently been characterized for mammalian reovirus [
24]. Alternatively, the different levels of plasma viremia could be differences in affinity and binding to erythrocytic surface molecules. Virus’ affinity for and binding to the erythrocyte surface has been known for several virus families, including the reoviruses [
25]. The infectious salmon anemia virus is an example of a salmonid virus that binds to the sialic acids on the erythrocyte surface [
26]. PRV-1 has a putative conserved sialic binding motif in the σ1 amino acid sequence [
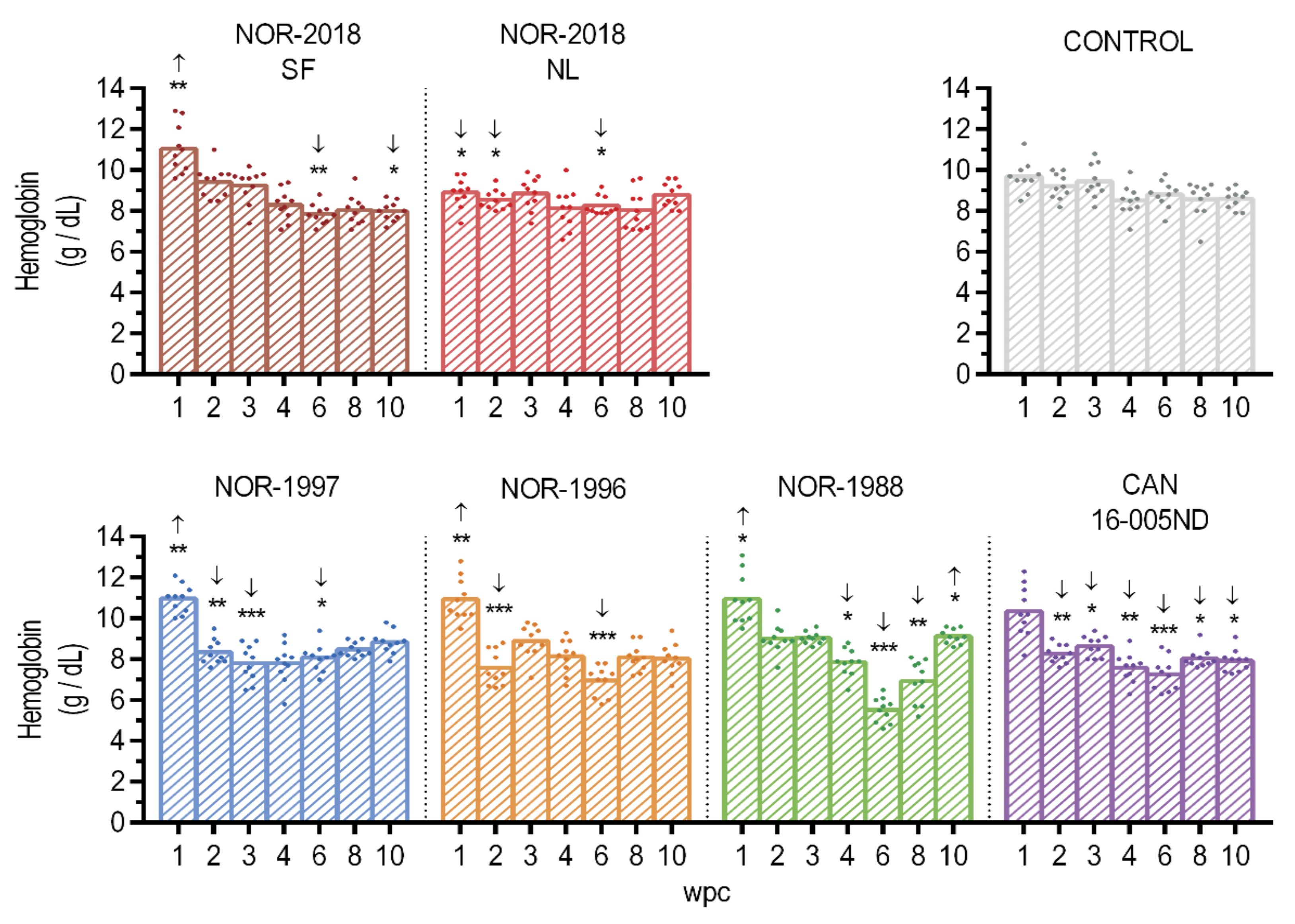
16]. Higher affinity to erythrocytes and thus more efficient removal of virus from plasma of the low virulent virus isolates could therefore, theoretically, explaining the differences in plasma viremia between the low- and high virulent groups. This is in accordance with the findings of presence of PRV-1 proteins in all erythrocytes in the low virulent groups but only in a fraction of the erythrocytes in the high virulent groups. Further characterization of the binding to and release from erythrocytes is needed to explain the differences in plasma viremia between high and low virulent isolates. It should be noted that at 6 wpc there was a substantial drop with little or no viral RNA in plasma. This was most pronounced in the Norwegian 2018 and Canadian isolate, thus not consistent with virulence differences. However, the results were surprising and difficult to put into a biological context. Although we did not reveal any quality issues with the samples collected at this timepoint, a non-biological explanation cannot be ruled out.
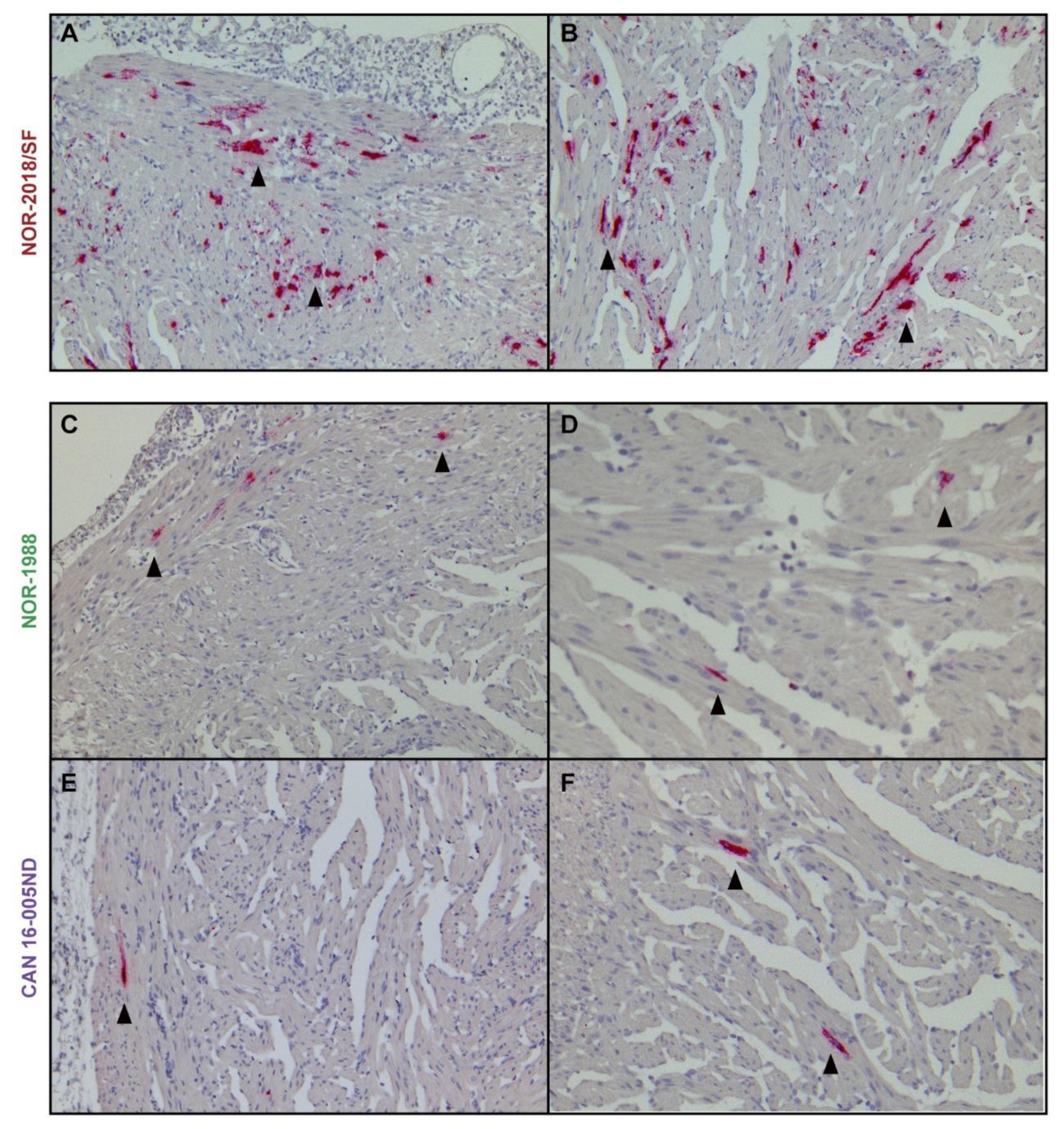
The ability to induce the heart lesions observed during HSMI, could be correlated to the level of plasma viremia due to more circulating virus available for infection of cardiomyocytes. In the high virulent groups, a high number of PRV positive cardiomyocytes was detected during development of HSMI, consistent with a previous study of a Norwegian field isolate [
11]. In comparison, lower amount of virus was detected in cardiomyocytes in the low virulent groups. The results of the in-situ hybridization were supported by the RT-qPCR of viral RNA. The difference in load could therefore be directly correlated to the level of plasma viremia. Alternatively, the high and low virulent isolates differ in their ability to enter or replicate in cardiomyocytes specifically. High loads of PRV in cardiomyocytes of infected fish have been observed in a reported outbreak in Canada [
6,
27]. It should be noted that the isolate of that outbreak, called B5690, is similar but not identical to the CAN 16-005ND isolate used in the present study [
12]. Furthermore, data from field material and a controlled challenge trial are not directly comparable.
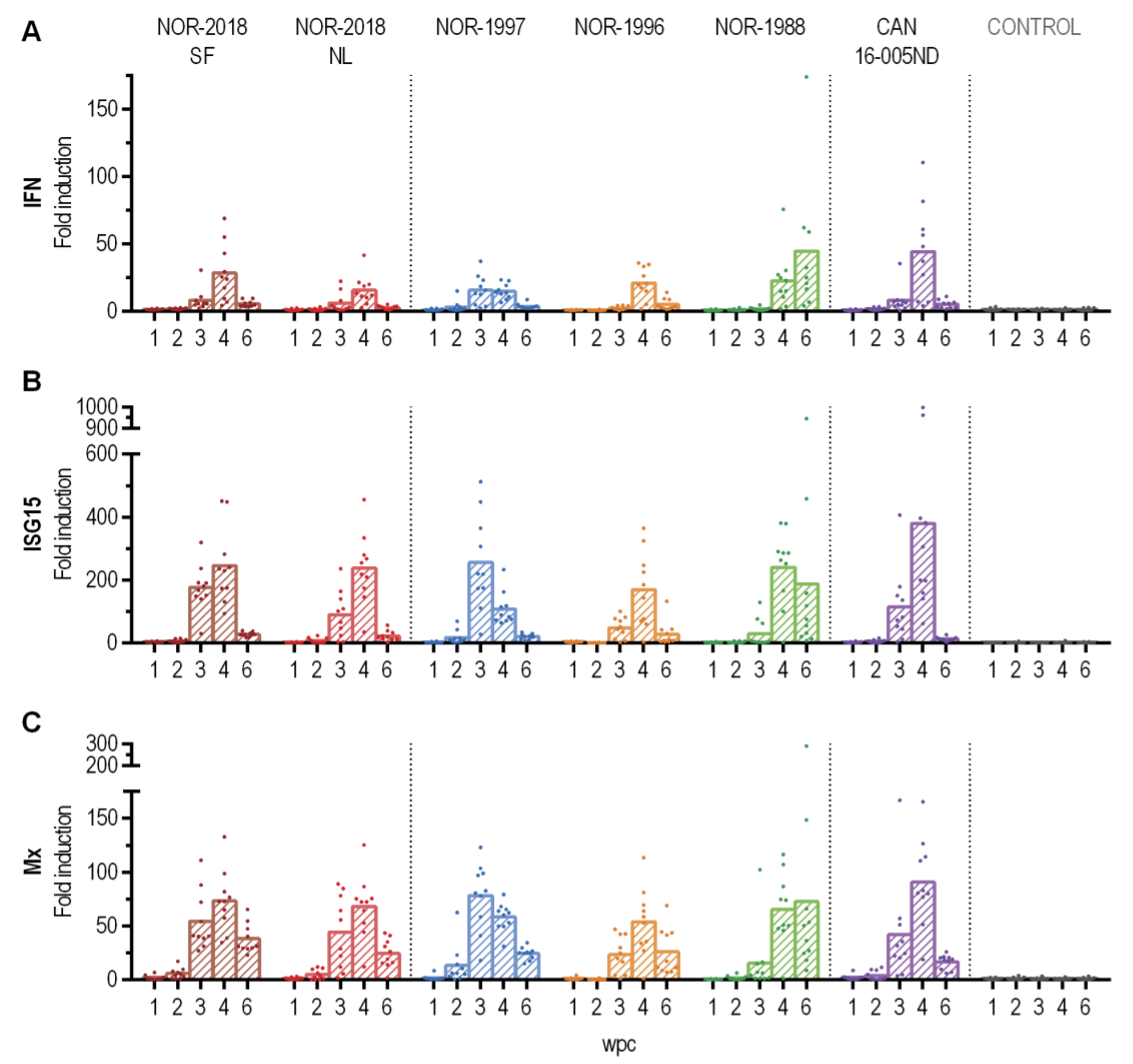
Differences in the innate antiviral response have been reported for PRV-1 infection in Atlantic salmon. Norwegian isolates have been reported to trigger a strong innate response in blood in multiple challenge trial and during ex vivo infection of erythrocytes [
2,
15,
28]. In contrast, infection with Canadian isolates have been reported with little or no immune response [
12,
13]. In the present study all isolates induced a strong innate antiviral response at similar levels with no differences that could be correlated to virulence. The reason for the previous deviating results is not known but could be attributed to host or technical factors. Furthermore, the differences in viral protein in blood and release of virus to plasma observed in the present study indicates other important virus-host interactions.
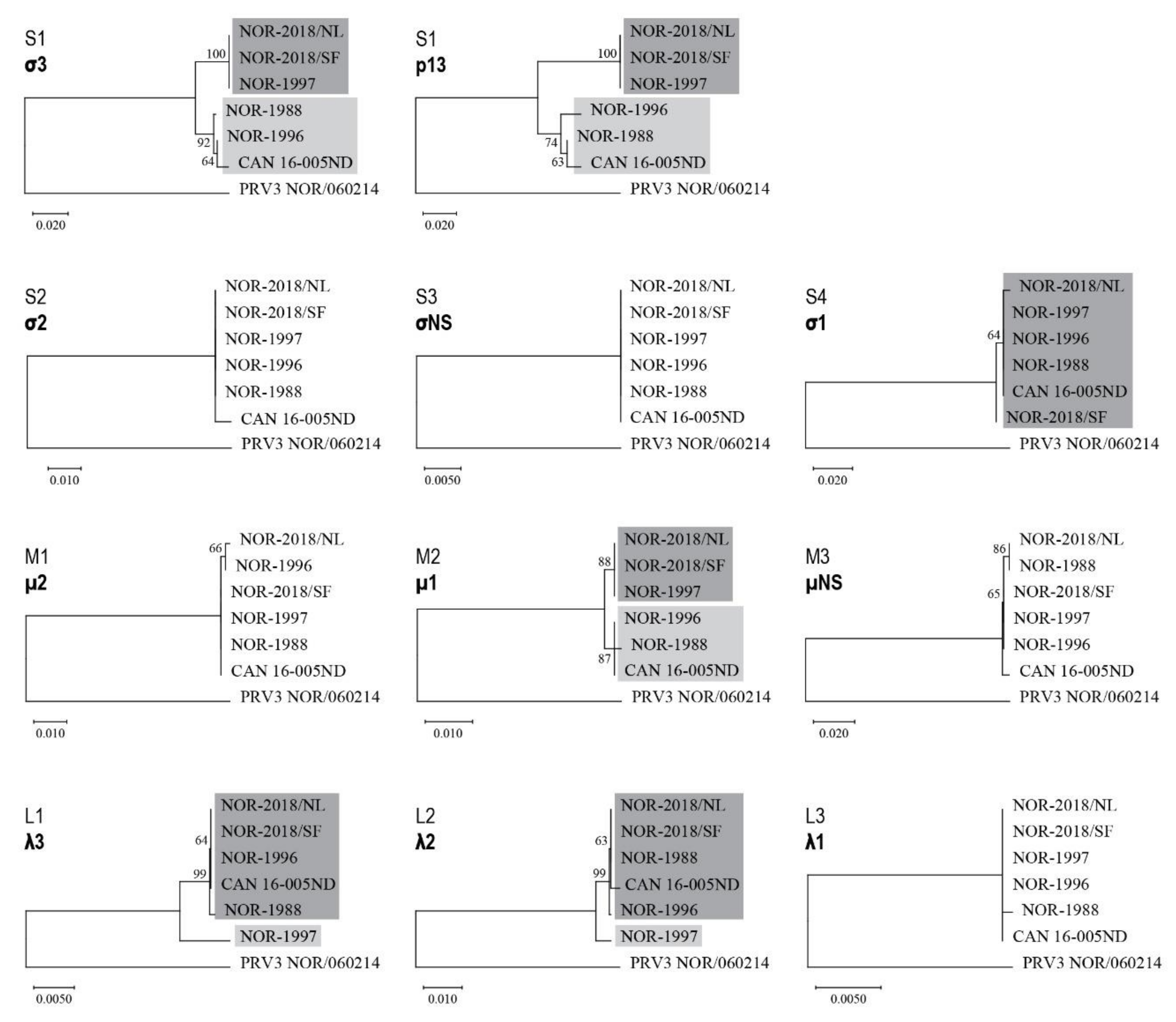
Sequence analysis of the high and low virulent isolates identified five segments (S1, M2, L1, L2, S4) which encode viral proteins that could be linked to phenotypical difference. Previous studies have reported evolution of segment S1 and M2 in Norway to be associated with the emergence of HSMI [
5], whereas these segments appear to have been relatively stable in isolates from North American Pacific Coast and the Faroe Islands with little or no HSMI [
5,
29]. In our study, the high virulent NOR-2018 isolates have the evolved variant of S1 and M2, whereas the low virulent Canadian isolate, NOR-1988 and NOR-1996 isolates cluster with the non-evolved variants. However, the NOR-1997 isolate was shown to be of low virulence but have S1 and M2 segments almost identical to that of the NOR-2018s. This excludes the possibility that the S1-M2 alone can explain the virulence differences. Interestingly, NOR-1997 has unique L1 and L2 segments, while these segments were almost identical in the other isolates. An assumed reassortment event combining the S1 and M2 segments of NOR-1997 with L1 and L2 segment of either NOR-1988 or NOR-1996 would in fact generate a virus with a genetic constellation similar to the virulent NOR-2018 isolates.
The segments (S1, M2, L1, L2, S4) and their encoded viral proteins (σ3, µ1, λ2, σ1, λ3 and p13) are potentially related to virulence but the underlying mechanism for development of HSMI were not clarified. The proteins include four structural proteins and two non-structural proteins. Interestingly, the structural proteins are all part of the outer capsid, including σ3 (S1), µ1 (M2), λ2 (L1) and σ1 (S4) [
16]. Each protein has specific functions during entry and replication but they are also structurally linked in the outer capsid [
30]. The function of the outer capsid proteins is balancing between the capsid stability and optimal attachment and capsid disassembly during cell entry [
31]. The increased size and number of salmon farms has substantially increased the number of available hosts. A segmented RNA virus such as PRV will explore the available sequence space through mutation and reassortment to optimize its fitness accordingly. Further investigations are needed to see if the change in outer capsid proteins is related to particle stability, affinity to erythrocytes or replicative potential.
In virulence investigations, the viral receptor binding protein is often given specific attention. These proteins are major determinants of cell tropism and therefore often also of virulence, which is the case for σ1 in mammalian reovirus (MRV) [
32]. In the present study, variation was observed in PRV-1 σ1 (S4). The historical Norwegian and the Canadian isolates had identical σ1 amino acid sequence, whereas both Norwegian 2018 isolates differed from these by one amino acid, which was located at different positions: V107A in NOR-2018/SF and D252N in NOR-2018/NL. If implicated in virulence, these two amino acid changes imposed a common phenotypic effect.
The isolate reported to cause cardiac lesions in field in BC Canada (B5690) has been reported to be similar but not identical to the CAN 16-005ND isolate used in the present study [
12]; the isolate contains the non-evolved variant of segment S1 and M2 and lack the unique sequence of NOR-1997 in segment L1 and L2. Interestingly, the Canadian B5690 isolate contains an amino acid difference in σ1 (segment S4) position 252 (D252E) [
6]. Furthermore, another Canadian isolate (CAN 16-011D) which failed to induce cardiac lesions in a challenge trial [
12] has the same V107A change in σ1 as the virulent NOR2018-SF (data not shown). The latter finding could indicate that this amino acid position is not related to virulence. Overall, the amino acid positions 107 and 252 in the σ1 protein appears to be variable but functional effects of these variations are not clear.
Sequence differences in the two non-structural proteins λ3 (L1) and p13 (S1) were identified comparing high and low virulent isolates. The λ3 protein is the RNA-dependent RNA polymerase and thus key to replication [
16]. The λ3 amino acid sequence was almost identical for most isolates, except for the unique sequence observed in NOR-1997 but a potential role in virulence is not obvious. The p13 protein has been shown to be a cytotoxic integral membrane protein [
33]. It is encoded by an internal ORF on the bicistronic S1 segment and thus the coding sequences of p13 and σ3 proteins are intrinsically linked. Several of the structural proteins of reoviruses counteract the innate antiviral activity during replication, including the outer structural protein σ3 and λ2 [
34]. However, the analysis of the innate response did not indicate any substantial differences between low-and high-virulent isolates.
The present study used a dose standardized challenge model to study virulence. It is important to state that the combination PRV-1 and its natural host Atlantic salmon were the focus of this study. Although the conclusions regarding virulence factors in other salmonid species or of other PRV subtypes cannot be superimposed, the setup could be used in future investigations for different salmonid species and other PRV subtypes. In the trial, fish were challenged with a relatively high dose (5 × 10
7 particles per fish). Previous dose response trials suggest that high and low dose affect the timing of infection but not the severity of lesions [
2]. In the trial we challenged the fish by injection to provide a synchronized infection of fish within the group, which also enabled a timed comparison between different groups. Cohabitation challenge would provide a natural route of infection and add confirmation to the results but a classical cohabitation trial would make synchronized infection between groups more difficult. Previous studies suggest that cohabitational challenge of PRV-1 induce similar or more severe lesions compared to injection challenge [
1,
2,
9]. Alternatively, a bath challenge using standardized dose of purified virus would provide synchronized infection by the natural route of transmission.
This report is the first confirmation of virulence differences between PRV-1 isolates seen as the ability to induce cardiac lesions of different severity in Atlantic salmon. The high virulent isolates were associated with higher plasma viremia during early replication in blood, hypothesized to be associated with the subsequent pathogenic potential in the heart. Multiple genomic segments were identified with sequence variation differentiating high and low virulent isolates.
4. Materials and Methods
4.1. PRV-1 Isolates Used in the Study
Six PRV-1 isolates were included in the study; two Norwegian isolates from 2018 (NOR-2018/SF, NOR-2018/NL), three historical Norwegian isolates (NOR-1988, NOR-1996, NOR-1997) and one Canadian isolate (CAN 16-005ND). The two Norwegian 2018 isolates were collected from sea sites in the Norwegian counties Sogn og Fjordane (NOR-2018-SF) and Nordland (NOR-2018/NL) in 2018. The selected isolates were considered to be widespread in farmed Atlantic salmon in Norway but were not collected from an HSMI outbreak. The three historical Norwegian isolates NOR-1988, NOR-1996 and NOR-1997 all originated from sea sites in Norway two, three and eleven years prior to the first description of HSMI. NOR-1988 and NOR-1997 have previously been sequenced and had been passaged once through Atlantic salmon [
5], whereas NOR-1996 was identified in an archived plasma sample. The Canadian isolate (CAN 16-005ND) had previously been characterized and failed to induce HSMI in two experimental trials in Canada [
12,
13]. Briefly, the isolate originated from a cohort of healthy Atlantic salmon in Vancouver Island, Canada, with no history of HSMI and had been passaged three times through Atlantic salmon in Canada [
12] and once in Norway.
4.2. Virus Propagation
To obtain fresh blood samples with high viral loads enabling viral purification, all six isolates were passaged once through 90 g Atlantic salmon (StofnFiskur Optimal strain). The trial was conducted at VESO Vikan aquatic research facility (Namsos, Norway) and had been approved by the Norwegian Food Safety Authority (NFDA) according to the European Union Directive 2010/63/EU for animal experiments (permit number 11251). The PRV-1 containing material available varied between the isolates and the following inoculums were prepared: NOR-2018/SF and NOR-2018/NL as heparinized blood diluted in Leibovitz’s L15 medium (Life Technologies, Grand Island, NY, USA) (Cq 19.3 and Cq 23.7 respectively), NOR-1988 as purified virus in PBS (Cq 18.1), NOR-1996 as archived plasma sample diluted in L15 medium (Cq 18.7), NOR-1997 and CAN 16-005ND as pelleted blood cells diluted in L15 medium (Cq 19.5 and Cq 22.6 respectively). The inoculums were administered as intraperitoneal (Norwegian 2018 isolates) or intramuscular (historical Norwegian and Canadian isolates) injection in 16 naïve fish per group, using 0.1 mL per fish. The fish were sampled weekly up to 5 wpc collecting heparinized blood samples which were centrifuged to obtain pelleted blood cells. The load of viral RNA and viral protein was monitored by RT-qPCR and flow cytometry respectively (details described below). One blood pellet sample with high viral load was selected for viral purification: NOR-2018/SF Cq 15.3, NOR-2018/NL Cq 15.7, NOR-1988 Cq 17.9, NOR-1996 Cq 15.7, NOR-1997 Cq 15.4 and CAN 16-005ND Cq 19.6. Complete data set of the propagation shown in
Supplementary Table S3.
4.3. Virus Purification
The selected blood pellet from each isolate (0.5 mL) was diluted 1:10 in homogenization buffer (10 mM TrisCl, 50 mM NaCl, 10 mM 2-mercaptoethanol) and sonicated on ice for 10 s at 25 kHz five times with 1 min rest in between. PRV-1 was purified from the blood homogenate and viral particles separated on a CsCl gradient as previously described [
2]. The bottom of the tube was punctured using a 21G needle, fractions collected by gravitational drop (0.5–1.0 mL fractions) and density determined by cross referencing the refractive index figure with those computed by Bruner and Vinograd, 1965 [
35]. For each isolate, the CsCl fraction with the buoyant density of PRV-1 (1.34 g/mL) [
2]) was injected into a Slide-A-Lyzer Dialysis Cassette (Thermo Fisher Scientific, Waltham, MA, USA) and dialyzed against Dulbecco’s PBS without calcium and magnesium (Sigma-Aldrich, St. Louis, MO, USA) for >1 h, >3 h and then >12 h. PRV-1 particles were verified in all batches by transmission electron microscopy (TEM) of negative stained samples as previously described [
2]. A filamentous material was also observed by TEM, it could not be identified and was similar in all batches of virus. The purified batches were stored with 15% glycerol at −80 °C until used.
4.4. Virus Quantification
The copy number of the bathes of purified virus was determined by absolute quantification RT-qPCR. RNA was isolated from a 10 μL sample, diluted in 130 μL Dulbecco’s phosphate buffered saline (DPBS), using QIAamp Viral RNA mini kit according to manufacturer’s instructions (Qiagen, Hilden, Germany), eluting RNA in 50 μL. The eluted template RNA was denatured at 95 °C for 5 min using 5 μL as input for the RT-qPCR reaction performed with Qiagen OneStep kit (Qiagen, Hilden, Germany) targeting PRV segment S1, run with triplicate samples (conditions described later under PRV RT-qPCR). Absolute quantification of the PRV-1 particles was performed using a standard curve made from in vitro transcripts of the PRV segment S1 open reading frame (ORF) prepared as previously described [
2]. An eight-point standard curve from 10-fold serial dilutions of transcripts, ranging from 10
8 to 10
1 transcripts per sample was run with the purified PRV batches. The batches of purified virus contained the following copy number: NOR-2018/SF 7.3 × 10
9 copies/mL, NOR-2018/NL 2.4 × 10
10 copies/mL, NOR-1988 4.0 × 10
9 copies/mL, NOR-1996 1.6 × 10
10 copies/mL, NOR-1997 8.1 × 10
10 copies/mL, CAN 16-005ND 2.8 × 10
9 copies/mL.
4.5. Sequencing and Genome Assembly
RNA was isolated from 200 μL of the batches of purified virus from all six isolates (except 100 μL for CAN 16-005ND) using a combination of Trizol LS (Life Technologies) and RNeasy Mini spin column (Qiagen, Hilden, Germany). In brief, the purified virus was mixed with Trizol LS, added chloroform, then separating the phases by centrifugation. The aqueous phase was collected and proceeded with the RNeasy Mini spin column (Qiagen, Hilden, Germany) as recommended by the manufacturer, eluting isolated RNA in 30 μL RNase-free water.
Next-Generation Sequencing was performed using the Ion Total RNA-Seq Kit v2 library preparation kits (Thermo Fisher Scientific) following the manufacturer’s recommendations. The library preparation was setup up on a Ion Chef system and the samples were sequenced on Ion S5™ System from (Thermo Fisher Scientific).
The sequence reads generated by next-generation sequencing were imported to CLC genomics workbench v10.0.1 (Qiagen, Hilden, Germany) and mapped to reference genome, Piscine orthoreovirus isolate NOR2012-V3621. Default mapping parameters, mismatch cost = 2, insertion cost = 3 and deletion cost = 3 were used. Consensus sequences were generated from the mapping for all the individual segments of the PRV-1 isolates. Total number of mapped reads and average coverage mapping of PRV isolates is shown in
Supplementary Table S4. A list of the PRV-1 isolates used in the present study and their accession numbers are shown in
Table 1.
4.6. Sequence and Phylogentic Analysis
Multiple sequence alignments were performed using MUSCLE [
36] and MEGA X software v10.1.7 (available from
www.megasoftware.net) [
37]. Phylogenetic analyses were performed with MEGA X using full-length sequences from all ten gene segments and the eleven proteins known to be encoded by PRV-1. Maximum likelihood (ML) was used to generate the phylogenetic trees from gene segment sequences and neighbor joining (NJ) for the amino acid sequences, using the best-fitting substitution models suggested by the program. PRV-3 NOR/060214 (MG253807–MG253816) was selected as outgroup in all trees. Bootstrap values were calculated from 1000 replicates and values above 70 were considered significant [
20].
4.7. Experimental Challenge Trial
An experimental challenge trial comparing the six PRV-1 isolates was conducted at VESO Vikan aquatic research facility (Namsos, Norway). The trial had been approved by the Norwegian Food Safety Authority (#11251) according to the European Union Directive 2010/63/EU for animal experiments. A total of 500 Atlantic salmon (StofnFiskur Optimal strain) with an initial average weight of 90 g was used. The fish population had been screened (10 fish) and found negative for ISAV, SAV, PRV-1, PMCV and IPNV by RT-qPCR. Prior to challenge, the fish were acclimatized for one week. The fish were kept in tanks supplied with seawater of 32‰ salinity (30–36‰) at 12 ± 1 °C with continuous light, fed according to standard procedures and observed minimum once per day. The fish were anesthetized by bath immersion in benzocaine chloride (2 ± 5 min, 0.5 g/10 L water) prior to handling and euthanized using a higher concentration of benzocaine chloride (1 g/5 L water).
The experiment included six groups with the PRV-1 isolates NOR-2018/SF, NOR-2018/NL, NOR-1988, NOR-1996, NOR-1997, CAN 16-005ND and one negative control group. In each group, 70 fish were challenge with an intramuscular injection of purified virus in PBS (100 uL), using a standardized dose of 5 × 107 particles per fish. The control group was injected with PBS.
At 1, 2, 3, 4, 6, 8 and 10 wpc, 10 fish were sampled from each group. Heparinized blood was collected from the caudal vein and the hemoglobin concentrations was immediately measured by HemoCue Hb 201+ (HemoCue, Ängelhom, Sweden) according to the manufacturer’s instructions. The blood cells and plasma were analyzed for PRV protein by flow cytometry and western blotting respectively. Blood was centrifuged at 3000 g for 5 min at 4 °C to separate blood cells and plasma which was analyzed for PRV RNA content by RT-qPCR. The blood cells and plasma were analyzed for PRV protein by flow cytometry and western blotting respectively. Tissue from a heart was sampled in RNAlater (Life Technologies, Carlsbad, CA, USA) for RT-qPCR analysis and parallel samples were harvested in 10% phosphate buffered formalin and used for histological analysis and in-situ hybridization.
4.8. Detection of Viral Protein by Flow Cytometry
Blood cells were analyzed for PRV-1 σ1-protein by flow cytometry. All steps were performed on ice. Heparinized blood were diluted 1:20 in flow buffer (PBS+ 1% BSA+ 0.05% azide), plated in 50 μL aliquots into 96-well plates and washed in flow buffer. The cells were fixed in IC Fixation Buffer (eBioscience, San Diago, CA, USA) and washed in Permeabilization Buffer (eBioscience, San Diago, CA, USA). The blood cells were stained for 30 min using polyclonal antibodies raised against PRV-1 σ1 with a 1:5000 dilution (Anti-σ1, #K275) [
9]. Finally, the cells were stained with secondary Alexa Fluor 488 conjugated anti-rabbit IgG (Molecular Probes, Eugene, Oregon, USA) (2 mg/mL diluted 1:800) for 30 min. As a negative control, parallel samples were run using the same procedure but without primary antibody. The stained samples were read on a Gallios Flow Cytometer (Beckman Coulter, Indianapolis, IN, USA) counting 30,000 cells per sample and the data were analyzed using the Kaluza software (Beckman Coulter). For each sample, the ΔMFI was calculated by subtracting the MFI of the negative control (without primary antibody) from the MFI of the sample with primary antibody.
4.9. RNA Isolation
From pelleted blood cells, total RNA was isolated from 20 μL samples, which was homogenized in QIAzol Lysis Reagent (Qiagen, Hilden, Germany) using 5 mm steel beads and TissueLyser II (Qiagen, Hilden, Germany) for 2 × 5 min at 25 Hz. Then added chloroform, centrifuged, collected the aqueous phase and proceeded with automated RNA isolation with the RNeasy Mini QIAcube Kit (Qiagen, Hilden, Germany) as described by the manufacturer. From heart samples, total RNA was isolated from 25 mg samples using RNeasy Mini QIAcube Kit (Qiagen, Hilden, Germany) as described by the manufacturer. The RNA was quantified using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific).
For plasma samples, total RNA was isolated from a 50 μL sample, diluted in 90 μL PBS, using QIAamp Viral RNA mini kit according to manufacturer’s instructions (Qiagen, Hilden, Germany) and eluting RNA in 50 μL.
4.10. Detection of Viral RNA by qPCR
Detection of PRV-1 RNA from blood cells, heart and plasma was performed by RT-qPCR targeting segment S1 using previously described primers and probe [
2]. For heart and blood cell samples, input RNA was set to 100 ng (5 μL of 20 ng/μL) of total RNA which had been denatured at 95 °C for 5 min. The RT-qPCR was performed using Brilliant III Ultra-Fast QRT-PCR Master Mix (Agilent, Santa Clara, CA, USA) according to the manufacturer’s instruction with 400 nM primer and 300 nM probe. The following cycle parameters was used: 10 min at 50 °C, 3 min at 95 °C, 35 cycles of 95 °C/5 s and 60 °C/ 10 s run in AriaMx (Agilent, Santa Clara, CA, USA). For the plasma samples, input RNA was set to 5 μL out of the 50 μL eluted RNA which had been denatured at 95 °C for 5 min. The RT-qPCR was performed using the Qiagen OneStep kit (Qiagen, Hilden, Germany) with the following conditions: 400 nM primer, 300 nM probe, 400 nM dNTPs, 1.26 mM MgCl2 and 1 × ROX reference dye. The following cycle parameters was used: 30 min at 50 °C, 15 min at 95 °C, 35 cycles of 94 °C/15 s, 60 °C/30 s and 72 °C/30 s in AriaMx (Agilent, Santa Clara, CA, USA). All samples from blood cells, heart and plasma were run in duplicate and a sample was defined as positive if both parallel samples had a Ct < 35.
4.11. Detection of Viral Protein by Western Blotting
Western blotting was performed to compare loads of PRV proteins in plasma (0.4 μL) at 3 and 4 wpc. The proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), using 4–12% Bis-Tris Criterion XT PreCast gels (Bio-Rad, Hercules, CA, USA). The proteins were transferred to a polyvinylidene fluoride membrane (Trans-Blot Rurbo Midi PVDF, Bio-Rad, Hercules, CA, USA) and incubated with primary antibody overnight at 4 °C using the following sera diluted 1:500; Anti-σ1 #K275 [
9], Anti-μ1C #K265 [
9], Anti-λ1 #K273 [
38] and Anti-σNS 01BO [
39]. After incubation with secondary antibody, anti-rabbit IgG-HRP (Amersham, GE Healthcare, Buchinghamshire, UK) (1:50,000), the PRV proteins were detected by chemiluminescence (Clarity Western ECL Substrate, Bio-Rad, Hercules, CA, USA). MagicMark (XP Western Protein Standard, Invitrogen) was used as ladder. The western blot was performed on plasma samples at 3 and 4 wpc, detecting σ1 in all groups and expanded to include μ1C, λ1 and σNS in group NOR-2018/SF and CAN 16-005ND.
4.12. Heart Histopathology Evaluation
Formalin fixed and paraffin embedded sections of heart tissue from the challenge experiment were stained with hematoxylin-eosin (HE). The slides from 3–10 wpc, including all six PRV-1 groups and the negative control group, were blinded for histopathological examination (
n = 350) and evaluated by one investigator. The scoring was determined by a visual analog scale (0 to 3) based on previously published criteria [
2], scoring each heart compartment separately (epicardium, compactum, spongiosum and atrium). The arithmetic mean was calculated for a total cardiac score.
4.13. In-Situ Hybridization
To compare the virus RNA load in the heart, heart sections from group NOR-2018/SF, NOR-1988 and CAN 16-005ND sampled at 4, 6 and 8 wpc were stained for PRV RNA by in-situ hybridization using RNAscope
® (RED) 2.5 HD Detection Kit (Advanced Cell Diagnostic, Newark, CA, USA) according to manufacturer’s instructions as previously described [
40]. In brief, the paraffin embedded tissue sections (5 µm) were dewaxed in ACD HybEZ™ II followed by hydrogen peroxide treatment, then boiled in RNAscope Target Retrieval Reagent and incubated with RNAscope Protease Plus in HybEZ™ oven. Each section was hybridized by RNAscope probe targeting PRV-1 genome segment L3 (Advanced Cell Diagnostics catalog number-537451) [
40]. Probe targeting Peptidylpropyl Isomerase B (PPIB) in Atlantic salmon (Advanced Cell Diagnostics, catalog number-494421) was used as reference target gene expression to test for RNA integrity in the samples. As the negative control, probe-DapB (Advanced Cell Diagnostics catalog number-310043) was used to evaluate cross reactivity. Fast Red chromogenic substrate was used for detection of signals amplified following manufacturer’s instructions. Counterstaining was done with 50% Gill’s hematoxylin solution and mounted with EcoMount (BioCare Medical, Pacheco, CA, USA).
4.14. Innate Immne Response
The gene expression of antiviral immune genes, including type I interferon (INFab), interferon-stimulated gene 15 (ISG 15) and myxovirus resistance GTPase (Mx1) in blood cells were analyzed at 1–6 wpc for all six PRV-1 groups and the control group. From each sample, 400 ng total RNA was reverse transcribed to cDNA using the QuantiTect Reverse Transcription Kit with gDNA wipeout buffer (Qiagen, Hilden, Germany). For qPCR, cDNA corresponding to 5 ng RNA was analyzed with Sso Advanced Universal SYBR Green Supermix (Bio-Rad, Hercules, CA, USA) and 10 µM of forward and reverse target-specific primers in a 10 µl volume in duplicate wells on a 384 well plate. Probes and primer sequences are given in
Supplementary Table S5. The amplification program (95 °C/15 s denaturation, 60 °C/30 s amplification) was run for 40 cycles in a CFX Touch Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA), followed by a melt point analysis. Results were analyzed using the software CFX Manager, version 3.1.1621.0826. The expression cycle threshold level was normalized against the elongation factor (EF) 1α reference gene (ΔCt) The ΔΔCt method was used to calculate relative expression levels and fold induction compared to samples from the uninfected control group from the same sampling point.
4.15. Statistical Analysis
Statistical comparison between different groups was performed using the non-parametric Mann-Whitney test due to the small sample size (n = 10). The comparison included PRV RNA load measured as Ct-values (blood cells, plasma and heart), hemoglobin concentration obtained by HemoCue, protein load in blood cells observed as MFI value by flow cytometry, histopathological heart scores and expression on innate immune genes. p-values ≤ 0.05 were considered as significant. All statistical analysis described were performed with GraphPad Prism (GraphPad Software Inc., La Jolla, CA, USA).

 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}