Putative Autoantigen Leiomodin-1 Is Expressed in the Human Brain and in the Membrane Fraction of Newly Formed Neurons

 , , and
, , and 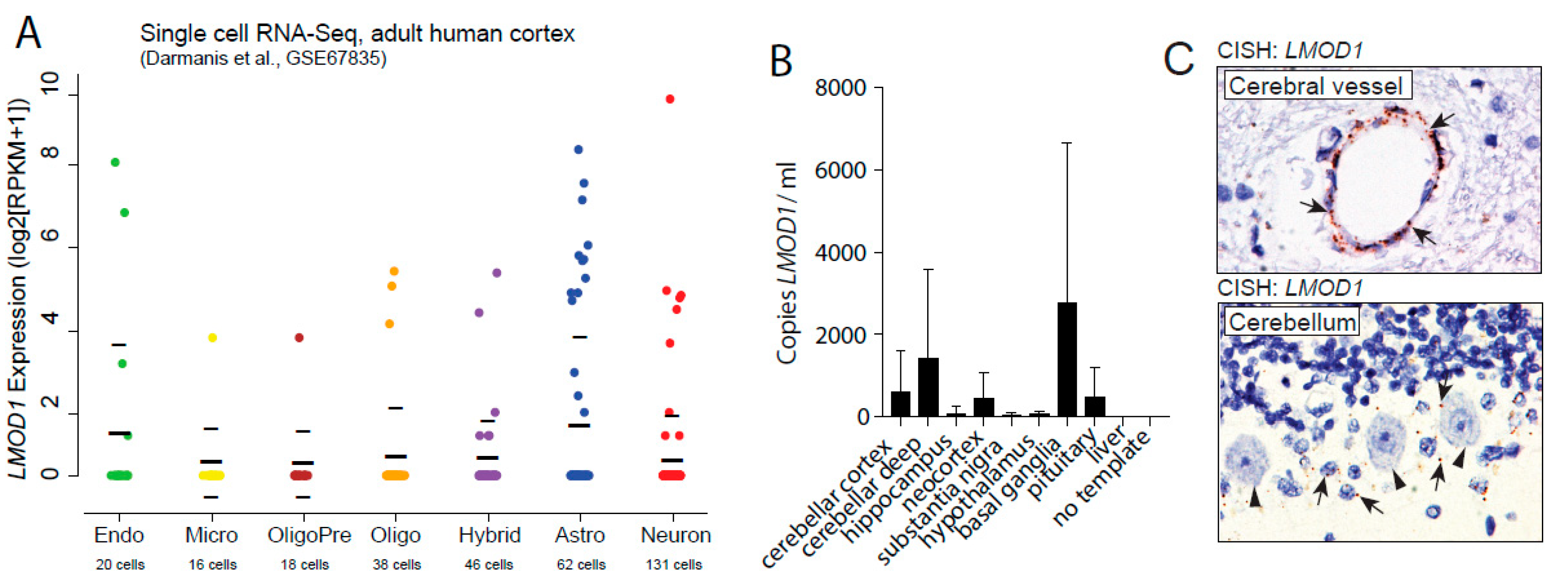{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
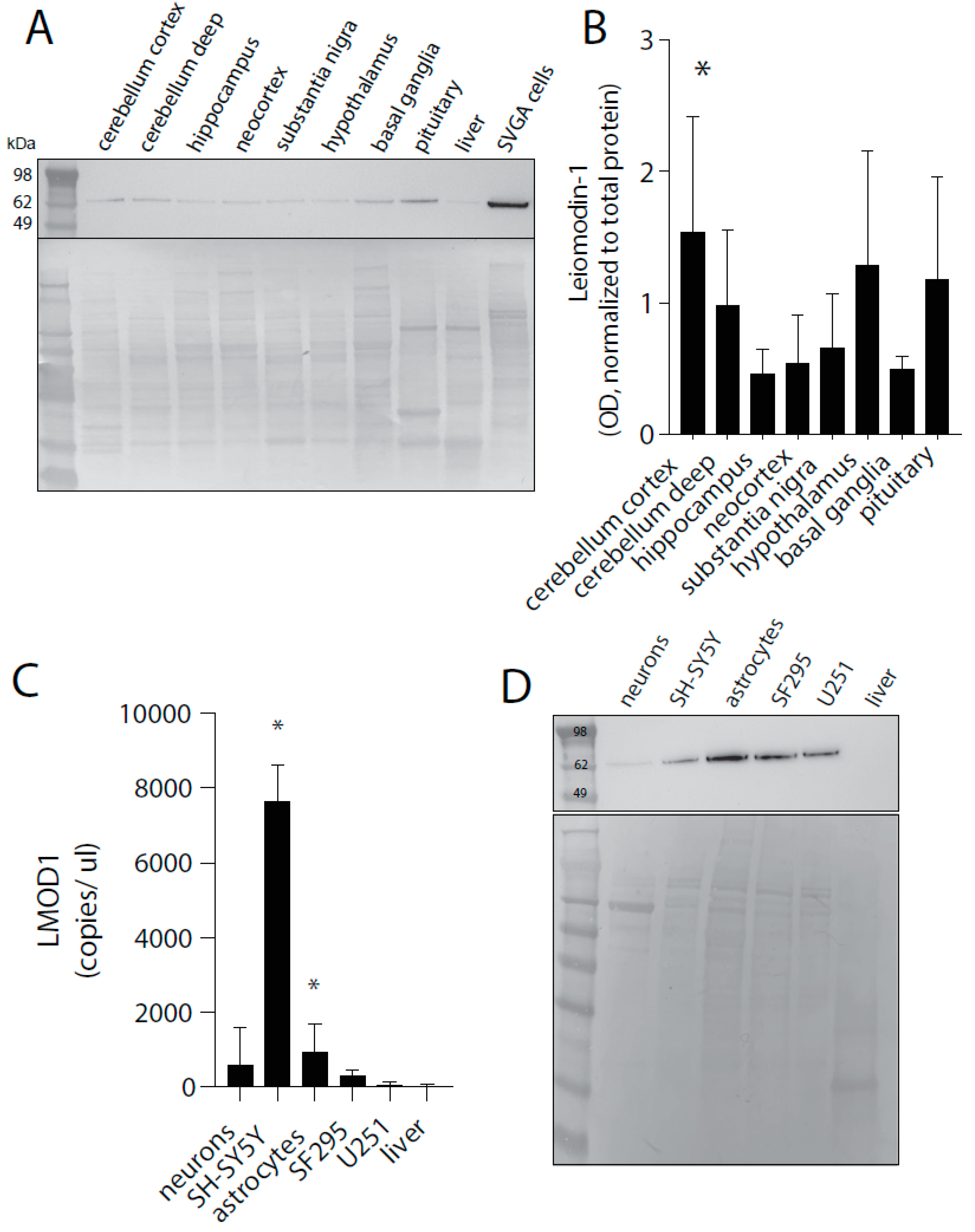
2.1. Leiomodin-1 Is Expressed in Neurons and Astrocytes of the Human CNS
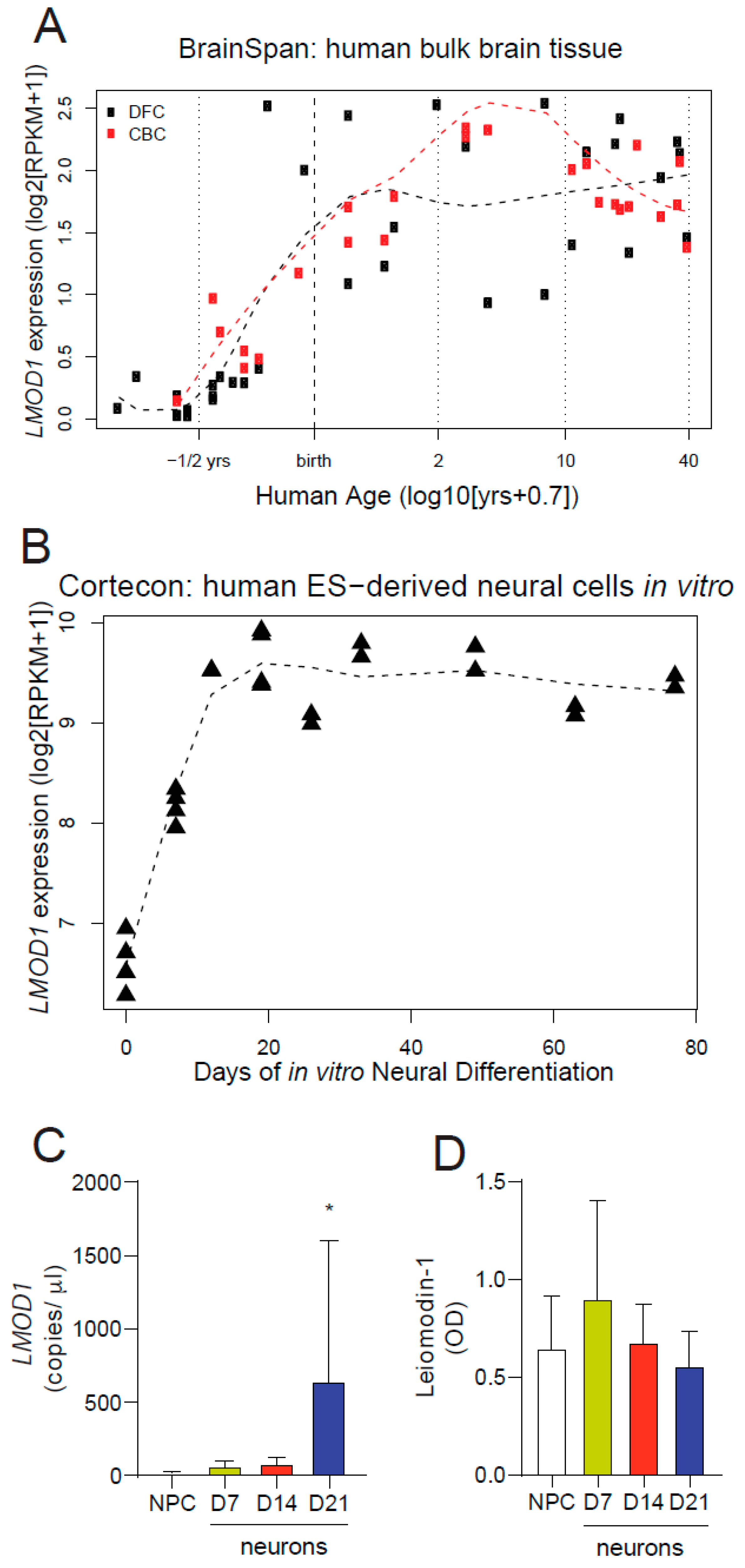
2.2. Leiomodin-1 Expression Changes during Development
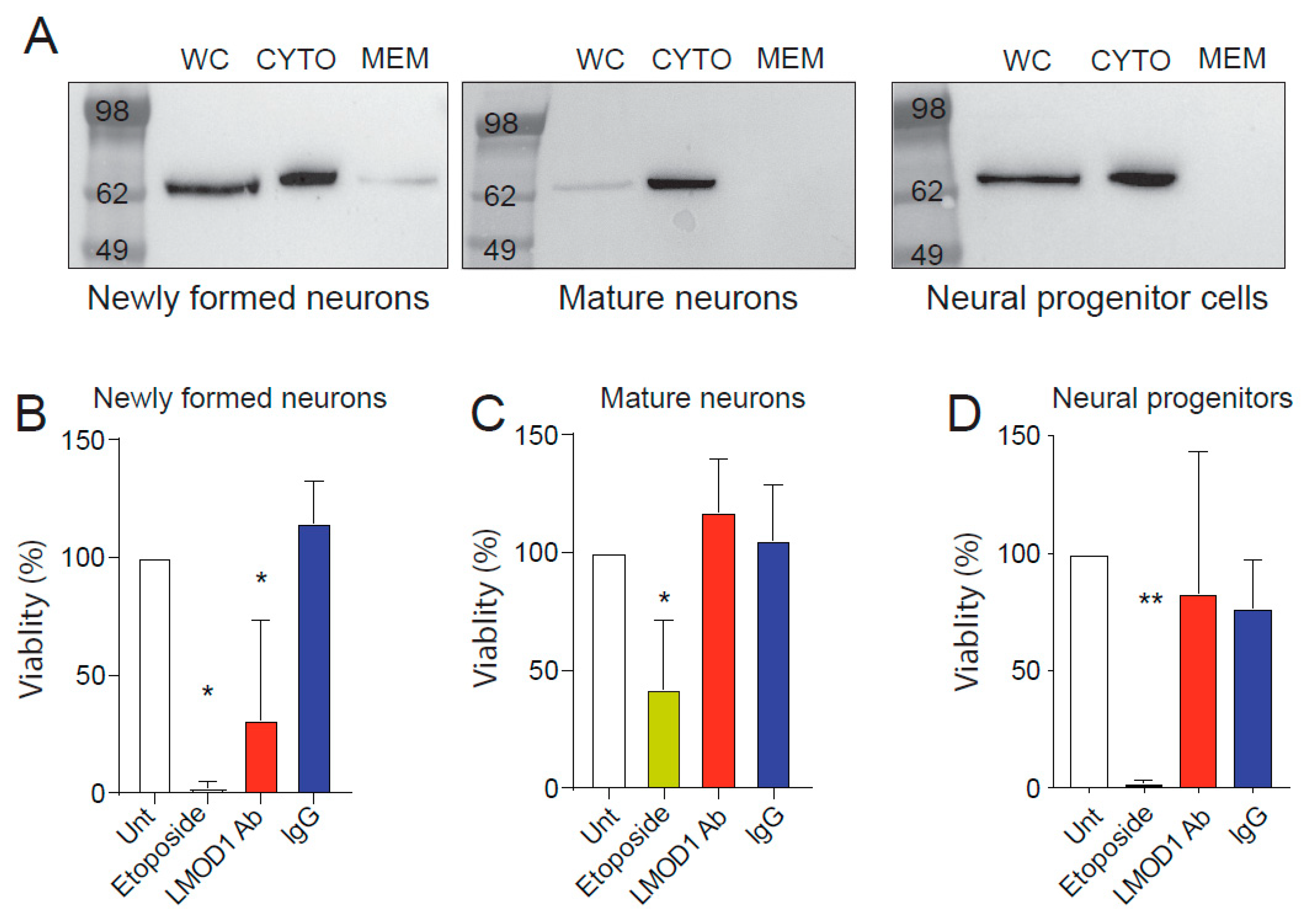
2.3. Leiomodin-1 Changes Subcellular Localization during Neural Development
2.4. Leiomodin-1 Antibodies Are Toxic to Newly Formed Neurons but Not to Mature Neurons
3. Discussion
4. Materials and Methods
4.1. In Vitro Neuronal and Glial Cell Differentiation
4.2. Cell Lines
4.3. Cell Fractionation
4.4. Ethical Approval
4.5. Immunoblotting
4.6. Immunohistochemistry
4.7. Immunofluorescence on Smooth Muscle Cells
4.8. RNA In Situ Hybridization
4.9. Quantitative PCR
4.10. Cellular Toxicity
4.11. In Silico Analysis
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dowell, S.F.; Sejvar, J.J.; Riek, L.; Vandemaele, K.A.; Lamunu, M.; Kuesel, A.C.; Schmutzhard, E.; Matuja, W.; Bunga, S.; Foltz, J.; et al. Nodding syndrome. Emerg. Infect. Dis. 2013, 19, 1374–1384. [Google Scholar] [CrossRef] [PubMed]
- Foltz, J.L.; Makumbi, I.; Sejvar, J.J.; Malimbo, M.; Ndyomugyenyi, R.; Atai-Omoruto, A.D.; Alexander, L.N.; Abang, B.; Melstrom, P.; Kakooza, A.M.; et al. An Epidemiologic Investigation of Potential Risk Factors for Nodding Syndrome in Kitgum District, Uganda. PLoS ONE 2013, 8, e66419. [Google Scholar] [CrossRef] [PubMed]
- Sejvar, J.J.; Kakooza, A.M.; Foltz, J.L.; Makumbi, I.; Atai-Omoruto, A.D.; Malimbo, M.; Ndyomugyenyi, R.; Alexander, L.N.; Abang, B.; Downing, R.G.; et al. Clinical, neurological, and electrophysiological features of nodding syndrome in Kitgum, Uganda: An observational case series. Lancet Neurol. 2013, 12, 166–174. [Google Scholar] [CrossRef]
- Idro, R.; Opoka, R.O.; Aanyu, H.T.; Kakooza-Mwesige, A.; Piloya-Were, T.; Namusoke, H.; Musoke, S.B.; Nalugya, J.; Bangirana, P.; Mwaka, A.D.; et al. Nodding syndrome in Ugandan children—Clinical features, brain imaging and complications: A case series. BMJ Open 2013, 3. [Google Scholar] [CrossRef]
- Winkler, A.; Friedrich, K.; Velicheti, S.; Dharsee, J.; Konig, R.; Nassri, A.; Meindl, M.; Kidunda, A.; Muller, T.; Jilek-Aall, L.; et al. MRI findings in people with epilepsy and nodding syndrome in an area endemic for onchocerciasis: An observational study. Afr. Health Sci. 2013, 13, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Winkler, A.S.; Friedrich, K.; Konig, R.; Meindl, M.; Helbok, R.; Unterberger, I.; Gotwald, T.; Dharsee, J.; Velicheti, S.; Kidunda, A.; et al. The head nodding syndrome--clinical classification and possible causes. Epilepsia 2008, 49, 2008–2015. [Google Scholar] [CrossRef] [PubMed]
- Konig, R.; Nassri, A.; Meindl, M.; Matuja, W.; Kidunda, A.R.; Siegmund, V.; Bretzel, G.; Loscher, T.; Jilek-Aall, L.; Schmutzhard, E.; et al. The role of Onchocerca volvulus in the development of epilepsy in a rural area of Tanzania. Parasitology 2010, 137, 1559–1568. [Google Scholar] [CrossRef] [PubMed]
- Tumwine, J.K.; Vandemaele, K.; Chungong, S.; Richer, M.; Anker, M.; Ayana, Y.; Opoka, M.L.; Klaucke, D.N.; Quarello, A.; Spencer, P.S. Clinical and epidemiologic characteristics of nodding syndrome in Mundri County, southern Sudan. Afr. Health Sci. 2012, 12, 242–248. [Google Scholar] [CrossRef]
- Chesnais, C.B.; Nana-Djeunga, H.C.; Njamnshi, A.K.; Lenou-Nanga, C.G.; Boulle, C.; Bissek, A.Z.; Kamgno, J.; Colebunders, R.; Boussinesq, M. The temporal relationship between onchocerciasis and epilepsy: A population-based cohort study. Lancet Infect. Dis. 2018, 18, 1278–1286. [Google Scholar] [CrossRef]
- Chesnais, C.B.; Bizet, C.; Campillo, J.T.; Njamnshi, W.Y.; Bopda, J.; Nwane, P.; Pion, S.D.; Njamnshi, A.K.; Boussinesq, M. A Second Population-Based Cohort Study in Cameroon Confirms the Temporal Relationship Between Onchocerciasis and Epilepsy. Open Forum Infect. Dis. 2020, 7, ofaa206. [Google Scholar] [CrossRef]
- Colebunders, R.; Hendy, A.; Mokili, J.L.; Wamala, J.F.; Kaducu, J.; Kur, L.; Tepage, F.; Mandro, M.; Mucinya, G.; Mambandu, G.; et al. Nodding syndrome and epilepsy in onchocerciasis endemic regions: Comparing preliminary observations from South Sudan and the Democratic Republic of the Congo with data from Uganda. BMC Res. Notes 2016, 9, 182. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.; Geerts, A.T.; Jol-van der Zijde, C.M.; Jacobson, L.; Lang, B.; Waters, P.; van Tol, M.J.; Stroink, H.; Neuteboom, R.F.; Brouwer, O.F.; et al. Neuronal antibodies in pediatric epilepsy: Clinical features and long-term outcomes of a historical cohort not treated with immunotherapy. Epilepsia 2016, 57, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.P.; Tyagi, R.; Lee, P.R.; Lee, M.H.; Johnson, K.R.; Kowalak, J.; Elkahloun, A.; Medynets, M.; Hategan, A.; Kubofcik, J.; et al. Nodding syndrome may be an autoimmune reaction to the parasitic worm Onchocerca volvulus. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Chereau, D.; Boczkowska, M.; Skwarek-Maruszewska, A.; Fujiwara, I.; Hayes, D.B.; Rebowski, G.; Lappalainen, P.; Pollard, T.D.; Dominguez, R. Leiomodin is an actin filament nucleator in muscle cells. Science 2008, 320, 239–243. [Google Scholar] [CrossRef]
- Darmanis, S.; Sloan, S.A.; Zhang, Y.; Enge, M.; Caneda, C.; Shuer, L.M.; Hayden Gephart, M.G.; Barres, B.A.; Quake, S.R. A survey of human brain transcriptome diversity at the single cell level. Proc. Natl. Acad. Sci. USA 2015, 112, 7285–7290. [Google Scholar] [CrossRef]
- Hawrylycz, M.J.; Lein, E.S.; Guillozet-Bongaarts, A.L.; Shen, E.H.; Ng, L.; Miller, J.A.; van de Lagemaat, L.N.; Smith, K.A.; Ebbert, A.; Riley, Z.L.; et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 2012, 489, 391–399. [Google Scholar] [CrossRef]
- Lein, E.S.; Hawrylycz, M.J.; Ao, N.; Ayres, M.; Bensinger, A.; Bernard, A.; Boe, A.F.; Boguski, M.S.; Brockway, K.S.; Byrnes, E.J.; et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007, 445, 168–176. [Google Scholar] [CrossRef]
- Spencer, P.S.; Mazumder, R.; Palmer, V.S.; Valdes Angues, R.; Pollanen, M.S. The etiology of nodding syndrome phenotypes remains unknown( section sign, section sign section sign). Rev. Neurol. 2020. [Google Scholar] [CrossRef]
- Bordeaux, J.; Welsh, A.; Agarwal, S.; Killiam, E.; Baquero, M.; Hanna, J.; Anagnostou, V.; Rimm, D. Antibody validation. Biotechniques 2010, 48, 197–209. [Google Scholar] [CrossRef]
- Sfanos, K.S.; Yegnasubramanian, S.; Nelson, W.G.; Lotan, T.L.; Kulac, I.; Hicks, J.L.; Zheng, Q.; Bieberich, C.J.; Haffner, M.C.; De Marzo, A.M. If this is true, what does it imply? How end-user antibody validation facilitates insights into biology and disease. Asian J. Urol. 2019, 6, 10–25. [Google Scholar] [CrossRef]
- Freedman, L.P.; Venugopalan, G.; Wisman, R. Reproducibility2020: Progress and priorities. F1000Res. 2017, 6, 604. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Sousa, A.M.M.; Gao, T.; Skarica, M.; Li, M.; Santpere, G.; Esteller-Cucala, P.; Juan, D.; Ferrandez-Peral, L.; Gulden, F.O.; et al. Spatiotemporal transcriptomic divergence across human and macaque brain development. Science 2018, 362. [Google Scholar] [CrossRef] [PubMed]
- van de Leemput, J.; Boles, N.C.; Kiehl, T.R.; Corneo, B.; Lederman, P.; Menon, V.; Lee, C.; Martinez, R.A.; Levi, B.P.; Thompson, C.L.; et al. CORTECON: A temporal transcriptome analysis of in vitro human cerebral cortex development from human embryonic stem cells. Neuron 2014, 83, 51–68. [Google Scholar] [CrossRef]
- Conley, C.A. Leiomodin and tropomodulin in smooth muscle. Am. J. Physiol. Cell Physiol. 2001, 280, C1645–C1656. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.P.; Sejvar, J.; Nutman, T.B.; Nath, A. The Pathogenesis of Nodding Syndrome. Annu. Rev. Pathol. 2020, 15, 395–417. [Google Scholar] [CrossRef] [PubMed]
- Echodu, R.; Edema, H.; Malinga, G.M.; Hendy, A.; Colebunders, R.; Moriku Kaducu, J.; Ovuga, E.; Haesaert, G. Is nodding syndrome in northern Uganda linked to consumption of mycotoxin contaminated food grains? BMC Res. Notes 2018, 11, 678. [Google Scholar] [CrossRef]
- Idro, R.; Opar, B.; Wamala, J.; Abbo, C.; Onzivua, S.; Mwaka, D.A.; Kakooza-Mwesige, A.; Mbonye, A.; Aceng, J.R. Is nodding syndrome an Onchocerca volvulus-induced neuroinflammatory disorder? Uganda’s story of research in understanding the disease. Int. J. Infect. Dis. 2016, 45, 112–117. [Google Scholar] [CrossRef]
- Pollanen, M.S.; Onzivua, S.; Robertson, J.; McKeever, P.M.; Olawa, F.; Kitara, D.L.; Fong, A. Nodding syndrome in Uganda is a tauopathy. Acta Neuropathol. 2018, 136, 691–697. [Google Scholar] [CrossRef]
- Idro, R.; Namusoke, H.; Abbo, C.; Mutamba, B.B.; Kakooza-Mwesige, A.; Opoka, R.O.; Musubire, A.K.; Mwaka, A.D.; Opar, B.T. Patients with nodding syndrome in Uganda improve with symptomatic treatment: A cross-sectional study. BMJ Open 2014, 4, e006476. [Google Scholar] [CrossRef]
- Boussinesq, M.; Pion, S.D.; Demanga, N.; Kamgno, J. Relationship between onchocerciasis and epilepsy: A matched case-control study in the Mbam Valley, Republic of Cameroon. Trans. R. Soc. Trop. Med. Hyg. 2002, 96, 537–541. [Google Scholar] [CrossRef]
- Kaiser, C.; Asaba, G.; Kasoro, S.; Rubaale, T.; Kabagambe, G.; Mbabazi, M. Mortality from epilepsy in an onchocerciasis-endemic area in West Uganda. Trans. R. Soc. Trop. Med. Hyg. 2007, 101, 48–55. [Google Scholar] [CrossRef]
- Kaiser, C.; Asaba, G.; Leichsenring, M.; Kabagambe, G. High incidence of epilepsy related to onchocerciasis in West Uganda. Epilepsy Res. 1998, 30, 247–251. [Google Scholar] [CrossRef]
- Kaiser, C.; Kipp, W.; Asaba, G.; Mugisa, C.; Kabagambe, G.; Rating, D.; Leichsenring, M. The prevalence of epilepsy follows the distribution of onchocerciasis in a west Ugandan focus. Bull. World Health Organ. 1996, 74, 361–367. [Google Scholar]
- Kaiser, C.; Pion, S.D.; Boussinesq, M. Case-control studies on the relationship between onchocerciasis and epilepsy: Systematic review and meta-analysis. PLoS Negl. Trop. Dis. 2013, 7, e2147. [Google Scholar] [CrossRef] [PubMed]
- Ngugi, A.K.; Bottomley, C.; Kleinschmidt, I.; Wagner, R.G.; Kakooza-Mwesige, A.; Ae-Ngibise, K.; Owusu-Agyei, S.; Masanja, H.; Kamuyu, G.; Odhiambo, R.; et al. Prevalence of active convulsive epilepsy in sub-Saharan Africa and associated risk factors: Cross-sectional and case-control studies. Lancet Neurol. 2013, 12, 253–263. [Google Scholar] [CrossRef]
- Pion, S.D.; Kaiser, C.; Boutros-Toni, F.; Cournil, A.; Taylor, M.M.; Meredith, S.E.; Stufe, A.; Bertocchi, I.; Kipp, W.; Preux, P.M.; et al. Epilepsy in onchocerciasis endemic areas: Systematic review and meta-analysis of population-based surveys. PLoS Negl. Trop. Dis. 2009, 3, e461. [Google Scholar] [CrossRef] [PubMed]
- Boczkowska, M.; Rebowski, G.; Kremneva, E.; Lappalainen, P.; Dominguez, R. How Leiomodin and Tropomodulin use a common fold for different actin assembly functions. Nat. Commun. 2015, 6, 8314. [Google Scholar] [CrossRef]
- Halim, D.; Wilson, M.P.; Oliver, D.; Brosens, E.; Verheij, J.B.; Han, Y.; Nanda, V.; Lyu, Q.; Doukas, M.; Stoop, H.; et al. Loss of LMOD1 impairs smooth muscle cytocontractility and causes megacystis microcolon intestinal hypoperistalsis syndrome in humans and mice. Proc. Natl. Acad. Sci. USA 2017, 114, E2739–E2747. [Google Scholar] [CrossRef]
- Conley, C.A.; Fritz-Six, K.L.; Almenar-Queralt, A.; Fowler, V.M. Leiomodins: Larger members of the tropomodulin (Tmod) gene family. Genomics 2001, 73, 127–139. [Google Scholar] [CrossRef]
- Nanda, V.; Miano, J.M. Leiomodin 1, a new serum response factor-dependent target gene expressed preferentially in differentiated smooth muscle cells. J. Biol. Chem. 2012, 287, 2459–2467. [Google Scholar] [CrossRef]
- Piloya-Were, T.; Odongkara-Mpora, B.; Namusoke, H.; Idro, R. Physical growth, puberty and hormones in adolescents with Nodding Syndrome; a pilot study. BMC Res. Notes 2014, 7, 858. [Google Scholar] [CrossRef] [PubMed]
- Spencer, P.S.; Mazumder, R.; Palmer, V.S.; Pollanen, M.S. Nodding syndrome phenotypes. Rev. Neurol. 2019, 175, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Spencer, P.S.; Schmutzhard, E.; Winkler, A.S. Nodding Syndrome in the Spotlight—Placing Recent Findings in Perspective. Trends Parasitol. 2017, 33, 490–492. [Google Scholar] [CrossRef]
- Sanai, N.; Nguyen, T.; Ihrie, R.A.; Mirzadeh, Z.; Tsai, H.H.; Wong, M.; Gupta, N.; Berger, M.S.; Huang, E.; Garcia-Verdugo, J.M.; et al. Corridors of migrating neurons in the human brain and their decline during infancy. Nature 2011, 478, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Sorrells, S.F.; Paredes, M.F.; Cebrian-Silla, A.; Sandoval, K.; Qi, D.; Kelley, K.W.; James, D.; Mayer, S.; Chang, J.; Auguste, K.I.; et al. Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature 2018, 555, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.O.; Lybrand, Z.R.; Ito, N.; Brulet, R.; Tafacory, F.; Zhang, L.; Good, L.; Ure, K.; Kernie, S.G.; Birnbaum, S.G.; et al. Aberrant hippocampal neurogenesis contributes to epilepsy and associated cognitive decline. Nat. Commun. 2015, 6, 6606. [Google Scholar] [CrossRef]
- Opara, K.N.; Fagbemi, B.O. Population dynamics of onchocerca volvulus microfilariae in human host after six years of drug control. J. Vector Borne Dis. 2008, 45, 29–37. [Google Scholar]
- Nwaorgu, O.C.; Okeibunor, J.C. Onchocerciasis in pre-primary school children in Nigeria: Lessons for onchocerciasis country control programme. Acta. Trop. 1999, 73, 211–215. [Google Scholar] [CrossRef]
- Oguttu, D.; Byamukama, E.; Katholi, C.R.; Habomugisha, P.; Nahabwe, C.; Ngabirano, M.; Hassan, H.K.; Lakwo, T.; Katabarwa, M.; Richards, F.O.; et al. Serosurveillance to monitor onchocerciasis elimination: The Ugandan experience. Am. J. Trop. Med. Hyg. 2014, 90, 339–345. [Google Scholar] [CrossRef]
- Hughes, E.G.; Peng, X.; Gleichman, A.J.; Lai, M.; Zhou, L.; Tsou, R.; Parsons, T.D.; Lynch, D.R.; Dalmau, J.; Balice-Gordon, R.J. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J. Neurosci. 2010, 30, 5866–5875. [Google Scholar] [CrossRef]
- Chen, Z.; Wu, D.; Wang, K.; Luo, B. Cognitive Function Recovery Pattern in Adult Patients With Severe Anti-N-Methyl-D-Aspartate Receptor Encephalitis: A Longitudinal Study. Front. Neurol. 2018, 9, 675. [Google Scholar] [CrossRef] [PubMed]
- Guasp, M.; Dalmau, J. Encephalitis associated with antibodies against the NMDA receptor. Med. Clin. 2018, 151, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, J.; Toes, R.E.; Huizinga, T.W.; van der Woude, D. The role of anticitrullinated protein antibodies in the early stages of rheumatoid arthritis. Curr. Opin. Rheumatol. 2016, 28, 275–281. [Google Scholar] [CrossRef]
- Insel, R.A.; Dunne, J.L.; Atkinson, M.A.; Chiang, J.L.; Dabelea, D.; Gottlieb, P.A.; Greenbaum, C.J.; Herold, K.C.; Krischer, J.P.; Lernmark, A.; et al. Staging presymptomatic type 1 diabetes: A scientific statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care 2015, 38, 1964–1974. [Google Scholar] [CrossRef]
- Arbuckle, M.R.; McClain, M.T.; Rubertone, M.V.; Scofield, R.H.; Dennis, G.J.; James, J.A.; Harley, J.B. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N. Engl. J. Med. 2003, 349, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Rosen, A.; Casciola-Rosen, L. Autoantigens as Partners in Initiation and Propagation of Autoimmune Rheumatic Diseases. Annu. Rev. Immunol. 2016, 34, 395–420. [Google Scholar] [CrossRef] [PubMed]
- Major, E.O.; Miller, A.E.; Mourrain, P.; Traub, R.G.; de Widt, E.; Sever, J. Establishment of a line of human fetal glial cells that supports JC virus multiplication. Proc. Natl. Acad. Sci. USA 1985, 82, 1257–1261. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Fan, Y.S.; Chow, L.H.; Van Den Diepstraten, C.; van Der Veer, E.; Sims, S.M.; Pickering, J.G. Innate diversity of adult human arterial smooth muscle cells: Cloning of distinct subtypes from the internal thoracic artery. Circ. Res. 2001, 89, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Baena-Del Valle, J.A.; Zheng, Q.; Hicks, J.L.; Fedor, H.; Trock, B.J.; Morrissey, C.; Corey, E.; Cornish, T.C.; Sfanos, K.S.; De Marzo, A.M. Rapid Loss of RNA Detection by In Situ Hybridization in Stored Tissue Blocks and Preservation by Cold Storage of Unstained Slides. Am. J. Clin. Pathol. 2017, 148, 398–415. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nauen, D.W.; Haffner, M.C.; Kim, J.; Zheng, Q.; Yin, H.; DeMarzo, A.M.; Mahairaki, V.; Colantuoni, C.; Pickering, J.G.; Johnson, T.P. Putative Autoantigen Leiomodin-1 Is Expressed in the Human Brain and in the Membrane Fraction of Newly Formed Neurons. Pathogens 2020, 9, 1036. https://doi.org/10.3390/pathogens9121036
Nauen DW, Haffner MC, Kim J, Zheng Q, Yin H, DeMarzo AM, Mahairaki V, Colantuoni C, Pickering JG, Johnson TP. Putative Autoantigen Leiomodin-1 Is Expressed in the Human Brain and in the Membrane Fraction of Newly Formed Neurons. Pathogens. 2020; 9(12):1036. https://doi.org/10.3390/pathogens9121036
Chicago/Turabian StyleNauen, David W., Michael C. Haffner, Juyun Kim, Qizhi Zheng, Hao Yin, Angelo M. DeMarzo, Vasiliki Mahairaki, Carlo Colantuoni, J. Geoffrey Pickering, and Tory P. Johnson. 2020. "Putative Autoantigen Leiomodin-1 Is Expressed in the Human Brain and in the Membrane Fraction of Newly Formed Neurons" Pathogens 9, no. 12: 1036. https://doi.org/10.3390/pathogens9121036
APA StyleNauen, D. W., Haffner, M. C., Kim, J., Zheng, Q., Yin, H., DeMarzo, A. M., Mahairaki, V., Colantuoni, C., Pickering, J. G., & Johnson, T. P. (2020). Putative Autoantigen Leiomodin-1 Is Expressed in the Human Brain and in the Membrane Fraction of Newly Formed Neurons. Pathogens, 9(12), 1036. https://doi.org/10.3390/pathogens9121036