Roles of the Non-Structural Proteins of Influenza A Virus


{kind=link}
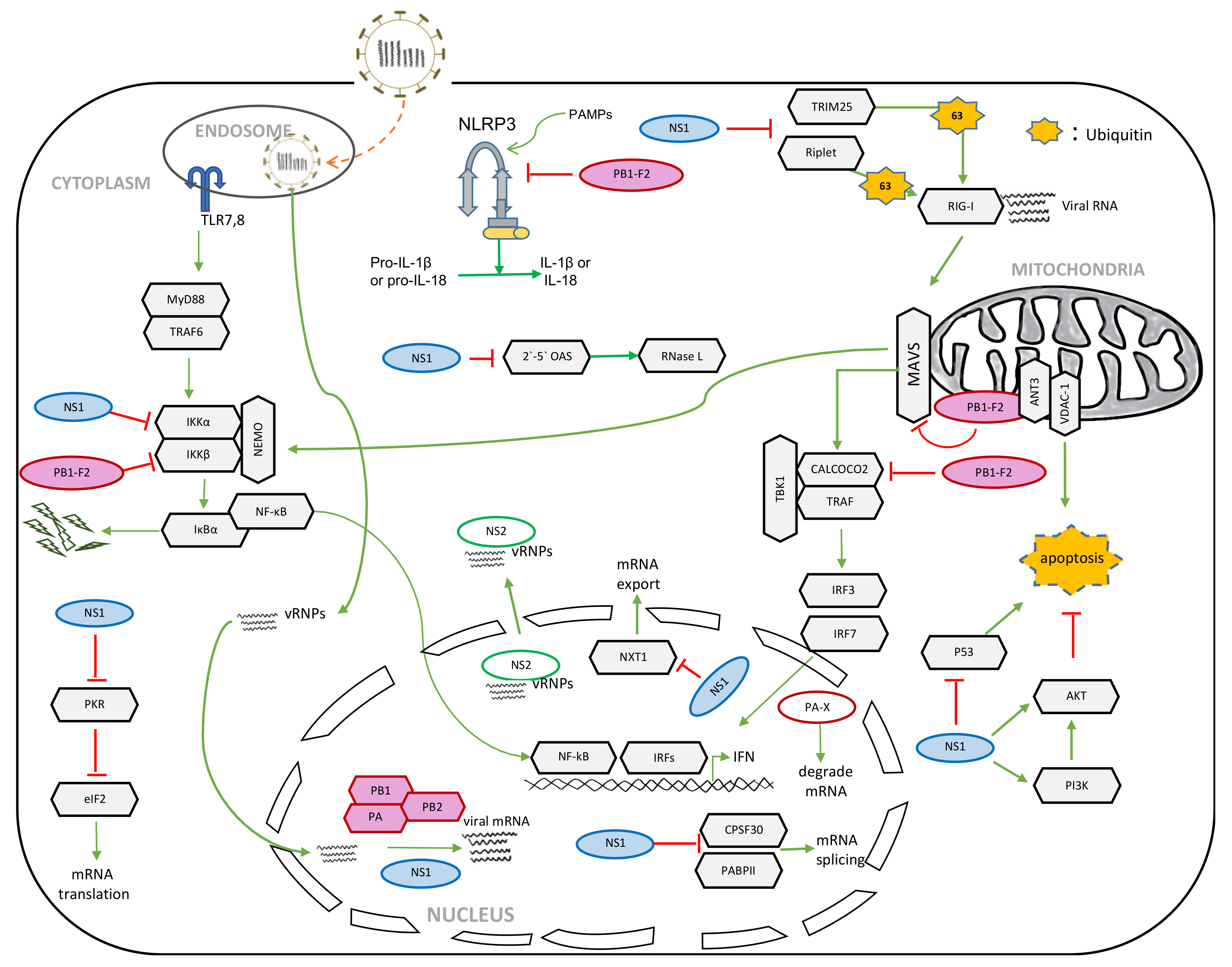{kind=link}
Abstract
1. Introduction
2. NS1
2.1. Role of NS1 in the Suppression of Host Innate Immunity
2.2. Role of NS1 in the Shutoff of Host Gene Expression
2.3. Role of NS1 in Apoptosis
2.4. Role of NS1 in Viral Replication
2.5. Virulence Determinant in NS1
3. NS2
3.1. Role of NS2 in vRNP Nuclear Export
3.2. Role of NS2 in Viral RNA Transcription and Replication
4. PB1-F2
4.1. Role of PB1-F2 in Apoptosis
4.2. Role of PB1-F2 in Host Innate Immune Responses
4.3. Role of PB1-F2 in Inflammasome Activation
4.4. Role of Cytosolic and Nuclear PB1-F2
4.5. Virulence Sites in PB1-F2
5. PA-X
5.1. Role of PA-X in the Shutoff of Host Gene Expression
5.2. Role of PA-X in the Pathogenesis
6. Other Non-Structural Proteins
6.1. PB1-N40
6.2. PA-N155 and PA-N182
6.3. M42
6.4. NS3
7. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Tscherne, D.M.; Garcia-Sastre, A. Virulence determinants of pandemic influenza viruses. J. Clin. Investig. 2011, 121, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, S.; Kawaoka, Y. The pathogenesis of influenza virus infections: The contributions of virus and host factors. Curr. Opin. Immunol. 2011, 23, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Hale, B.G.; Albrecht, R.A.; Garcia-Sastre, A. Innate immune evasion strategies of influenza viruses. Future Microbiol. 2010, 5, 23–41. [Google Scholar] [CrossRef] [PubMed]
- Kawaoka, J. Influenza Virology: Current Topics; Caister Academic Press: Wymondham, UK, 2006. [Google Scholar]
- Lamb, R.A.; Krug, R.M. Orthomyxoviridae: The Viruses and Their Replication; Lippincott-Raven: Philadelphia, PA, USA, 1996; p. 63. [Google Scholar]
- Krammer, F.; Smith, G.J.D.; Fouchier, R.A.M.; Peiris, M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G.; et al. Influenza. Nat. Rev. Dis. Primers 2018, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Jagger, B.W.; Wise, H.M.; Kash, J.C.; Walters, K.A.; Wills, N.M.; Xiao, Y.L.; Dunfee, R.L.; Schwartzman, L.M.; Ozinsky, A.; Bell, G.L.; et al. An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science 2012, 337, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Wise, H.M.; Foeglein, A.; Sun, J.; Dalton, R.M.; Patel, S.; Howard, W.; Anderson, E.C.; Barclay, W.S.; Digard, P. A complicated message: Identification of a novel PB1-related protein translated from influenza A virus segment 2 mRNA. J. Virol. 2009, 83, 8021–8031. [Google Scholar] [CrossRef]
- Muramoto, Y.; Noda, T.; Kawakami, E.; Akkina, R.; Kawaoka, Y. Identification of novel influenza A virus proteins translated from PA mRNA. J. Virol. 2013, 87, 2455–2462. [Google Scholar] [CrossRef]
- Wise, H.M.; Hutchinson, E.C.; Jagger, B.W.; Stuart, A.D.; Kang, Z.H.; Robb, N.; Schwartzman, L.M.; Kash, J.C.; Fodor, E.; Firth, A.E.; et al. Identification of a novel splice variant form of the influenza A virus M2 ion channel with an antigenically distinct ectodomain. PLoS Pathog. 2012, 8, e1002998. [Google Scholar] [CrossRef]
- Selman, M.; Dankar, S.K.; Forbes, N.E.; Jia, J.J.; Brown, E.G. Adaptive mutation in influenza A virus non-structural gene is linked to host switching and induces a novel protein by alternative splicing. Emerg. Microbes Infect. 2012, 1, e42. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Wilkins, C.; Gale, M., Jr. Recognition of viruses by cytoplasmic sensors. Curr. Opin. Immunol. 2010, 22, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Naslund, T.I.; Liljestrom, P.; Weber, F.; Reis e Sousa, C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5’-phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzozka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; et al. 5’-Triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef]
- Kang, D.C.; Gopalkrishnan, R.V.; Wu, Q.; Jankowsky, E.; Pyle, A.M.; Fisher, P.B. mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc. Natl. Acad. Sci. USA 2002, 99, 637–642. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef]
- Cui, S.; Eisenacher, K.; Kirchhofer, A.; Brzozka, K.; Lammens, A.; Lammens, K.; Fujita, T.; Conzelmann, K.K.; Krug, A.; Hopfner, K.P. The C-terminal regulatory domain is the RNA 5’-triphosphate sensor of RIG-I. Mol. Cell 2008, 29, 169–179. [Google Scholar] [CrossRef]
- Oshiumi, H.; Matsumoto, M.; Hatakeyama, S.; Seya, T. Riplet/RNF135, a RING finger protein, ubiquitinates RIG-I to promote interferon-beta induction during the early phase of viral infection. J. Biol. Chem. 2009, 284, 807–817. [Google Scholar] [CrossRef]
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef]
- Ablasser, A.; Hur, S. Regulation of cGAS- and RLR-mediated immunity to nucleic acids. Nat. Immunol. 2020, 21, 17–29. [Google Scholar] [CrossRef]
- Garcia-Sastre, A.; Egorov, A.; Matassov, D.; Brandt, S.; Levy, D.E.; Durbin, J.E.; Palese, P.; Muster, T. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 1998, 252, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Talon, J.; Horvath, C.M.; Polley, R.; Basler, C.F.; Muster, T.; Palese, P.; Garcia-Sastre, A. Activation of interferon regulatory factor 3 is inhibited by the influenza A virus NS1 protein. J. Virol. 2000, 74, 7989–7996. [Google Scholar] [CrossRef] [PubMed]
- Rajsbaum, R.; Albrecht, R.A.; Wang, M.K.; Maharaj, N.P.; Versteeg, G.A.; Nistal-Villan, E.; Garcia-Sastre, A.; Gack, M.U. Species-specific inhibition of RIG-I ubiquitination and IFN induction by the influenza A virus NS1 protein. PLoS Pathog. 2012, 8, e1003059. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.S.; Huang, I.C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; Garcia-Sastre, A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe. 2009, 5, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Kuo, R.L.; Zhao, C.; Malur, M.; Krug, R.M. Influenza A virus strains that circulate in humans differ in the ability of their NS1 proteins to block the activation of IRF3 and interferon-beta transcription. Virology 2010, 408, 146–158. [Google Scholar] [CrossRef]
- Gao, S.; Song, L.; Li, J.; Zhang, Z.; Peng, H.; Jiang, W.; Wang, Q.; Kang, T.; Chen, S.; Huang, W. Influenza A virus-encoded NS1 virulence factor protein inhibits innate immune response by targeting IKK. Cell Microbiol. 2012, 14, 1849–1866. [Google Scholar] [CrossRef]
- Min, J.Y.; Krug, R.M. The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: Inhibiting the 2’-5’ oligo (A) synthetase/RNase L pathway. Proc. Natl. Acad. Sci. USA 2006, 103, 7100–7105. [Google Scholar] [CrossRef]
- Nemeroff, M.E.; Barabino, S.M.; Li, Y.; Keller, W.; Krug, R.M. Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3’end formation of cellular pre-mRNAs. Mol. Cell 1998, 1, 991–1000. [Google Scholar] [CrossRef]
- Noah, D.L.; Twu, K.Y.; Krug, R.M. Cellular antiviral responses against influenza A virus are countered at the posttranscriptional level by the viral NS1A protein via its binding to a cellular protein required for the 3’ end processing of cellular pre-mRNAS. Virology 2003, 307, 386–395. [Google Scholar] [CrossRef]
- Twu, K.Y.; Noah, D.L.; Rao, P.; Kuo, R.L.; Krug, R.M. The CPSF30 binding site on the NS1A protein of influenza A virus is a potential antiviral target. J. Virol. 2006, 80, 3957–3965. [Google Scholar] [CrossRef]
- Das, K.; Ma, L.C.; Xiao, R.; Radvansky, B.; Aramini, J.; Zhao, L.; Marklund, J.; Kuo, R.L.; Twu, K.Y.; Arnold, E.; et al. Structural basis for suppression of a host antiviral response by influenza A virus. Proc. Natl. Acad. Sci. USA 2008, 105, 13093–13098. [Google Scholar] [CrossRef] [PubMed]
- Hale, B.G.; Steel, J.; Medina, R.A.; Manicassamy, B.; Ye, J.; Hickman, D.; Hai, R.; Schmolke, M.; Lowen, A.C.; Perez, D.R.; et al. Inefficient control of host gene expression by the 2009 pandemic H1N1 influenza A virus NS1 protein. J. Virol. 2010, 84, 6909–6922. [Google Scholar] [CrossRef] [PubMed]
- Twu, K.Y.; Kuo, R.L.; Marklund, J.; Krug, R.M. The H5N1 influenza virus NS genes selected after 1998 enhance virus replication in mammalian cells. J. Virol. 2007, 81, 8112–8121. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Fu, B.; Li, W.; Patil, G.; Liu, L.; Dorf, M.E.; Li, S. Comparative influenza protein interactomes identify the role of plakophilin 2 in virus restriction. Nat. Commun. 2017, 8, 13876. [Google Scholar] [CrossRef] [PubMed]
- Spesock, A.; Malur, M.; Hossain, M.J.; Chen, L.M.; Njaa, B.L.; Davis, C.T.; Lipatov, A.S.; York, I.A.; Krug, R.M.; Donis, R.O. The virulence of 1997 H5N1 influenza viruses in the mouse model is increased by correcting a defect in their NS1 proteins. J. Virol. 2011, 85, 7048–7058. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, Y.; Krug, R.M. Influenza A virus NS1 protein targets poly(A)-binding protein II of the cellular 3’-end processing machinery. Embo J. 1999, 18, 2273–2283. [Google Scholar] [CrossRef]
- Satterly, N.; Tsai, P.L.; van Deursen, J.; Nussenzveig, D.R.; Wang, Y.; Faria, P.A.; Levay, A.; Levy, D.E.; Fontoura, B.M. Influenza virus targets the mRNA export machinery and the nuclear pore complex. Proc. Natl. Acad. Sci. USA 2007, 104, 1853–1858. [Google Scholar] [CrossRef]
- Marazzi, I.; Ho, J.S.; Kim, J.; Manicassamy, B.; Dewell, S.; Albrecht, R.A.; Seibert, C.W.; Schaefer, U.; Jeffrey, K.L.; Prinjha, R.K.; et al. Suppression of the antiviral response by an influenza histone mimic. Nature 2012, 483, 428–433. [Google Scholar] [CrossRef]
- Min, J.Y.; Li, S.; Sen, G.C.; Krug, R.M. A site on the influenza A virus NS1 protein mediates both inhibition of PKR activation and temporal regulation of viral RNA synthesis. Virology 2007, 363, 236–243. [Google Scholar] [CrossRef]
- Li, S.; Min, J.Y.; Krug, R.M.; Sen, G.C. Binding of the influenza A virus NS1 protein to PKR mediates the inhibition of its activation by either PACT or double-stranded RNA. Virology 2006, 349, 13–21. [Google Scholar] [CrossRef]
- Tan, S.L.; Katze, M.G. Biochemical and genetic evidence for complex formation between the influenza A virus NS1 protein and the interferon-induced PKR protein kinase. J. Interferon Cytokine Res. 1998, 18, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Tawaratsumida, K.; Phan, V.; Hrincius, E.R.; High, A.A.; Webby, R.; Redecke, V.; Hacker, H. Quantitative proteomic analysis of the influenza A virus nonstructural proteins NS1 and NS2 during natural cell infection identifies PACT as an NS1 target protein and antiviral host factor. J. Virol. 2014, 88, 9038–9048. [Google Scholar] [CrossRef]
- Li, T.; Li, X.; Zhu, W.F.; Wang, H.Y.; Mei, L.; Wu, S.Q.; Lin, X.M.; Han, X.Q. NF90 is a novel influenza A virus NS1-interacting protein that antagonizes the inhibitory role of NS1 on PKR phosphorylation. FEBS Lett. 2016, 590, 2797–2810. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Macken, C.A.; Li, C.; Ozawa, M.; Goto, H.; Iswahyudi, N.F.; Nidom, C.A.; Chen, H.; Neumann, G.; Kawaoka, Y. Synergistic effect of the PDZ and p85beta-binding domains of the NS1 protein on virulence of an avian H5N1 influenza A virus. J. Virol. 2013, 87, 4861–4871. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.K.; Li, Y.; Liu, Q.; Anderson, D.H.; Babiuk, L.A.; Zhou, Y. SH3 binding motif 1 in influenza A virus NS1 protein is essential for PI3K/Akt signaling pathway activation. J. Virol. 2007, 81, 12730–12739. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, L.S.; Kazlauskas, A.; Melen, K.; Wagner, R.; Ziegler, T.; Julkunen, I.; Saksela, K. Avian and 1918 Spanish influenza a virus NS1 proteins bind to Crk/CrkL Src homology 3 domains to activate host cell signaling. J. Biol. Chem. 2008, 283, 5719–5727. [Google Scholar] [CrossRef] [PubMed]
- Hale, B.G.; Batty, I.H.; Downes, C.P.; Randall, R.E. Binding of influenza A virus NS1 protein to the inter-SH2 domain of p85 suggests a novel mechanism for phosphoinositide 3-kinase activation. J. Biol. Chem. 2008, 283, 1372–1380. [Google Scholar] [CrossRef]
- Hale, B.G.; Jackson, D.; Chen, Y.H.; Lamb, R.A.; Randall, R.E. Influenza A virus NS1 protein binds p85beta and activates phosphatidylinositol-3-kinase signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 14194–14199. [Google Scholar] [CrossRef]
- Ehrhardt, C.; Wolff, T.; Pleschka, S.; Planz, O.; Beermann, W.; Bode, J.G.; Schmolke, M.; Ludwig, S. Influenza A virus NS1 protein activates the PI3K/Akt pathway to mediate antiapoptotic signaling responses. J. Virol. 2007, 81, 3058–3067. [Google Scholar] [CrossRef]
- Zhirnov, O.P.; Konakova, T.E.; Wolff, T.; Klenk, H.D. NS1 protein of influenza A virus down-regulates apoptosis. J. Virol. 2002, 76, 1617–1625. [Google Scholar] [CrossRef]
- Shin, Y.K.; Liu, Q.; Tikoo, S.K.; Babiuk, L.A.; Zhou, Y. Influenza A virus NS1 protein activates the phosphatidylinositol 3-kinase (PI3K)/Akt pathway by direct interaction with the p85 subunit of PI3K. J. Gen. Virol. 2007, 88, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, D.B.; Mukatira, S.; Mehta, P.K.; Obenauer, J.C.; Su, X.; Webster, R.G.; Naeve, C.W. Persistent host markers in pandemic and H5N1 influenza viruses. J. Virol. 2007, 81, 10292–10299. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, M.; Nishihara, H.; Hasegawa, H.; Tashiro, M.; Wang, L.; Kimura, T.; Tanino, M.; Tsuda, M.; Tanaka, S. NS1-binding protein abrogates the elevation of cell viability by the influenza A virus NS1 protein in association with CRKL. Biochem. Biophys. Res. Commun. 2013, 441, 953–957. [Google Scholar] [CrossRef] [PubMed]
- Ayllon, J.; Garcia-Sastre, A.; Hale, B.G. Influenza A viruses and PI3K: Are there time, place and manner restrictions? Virulence 2012, 3, 411–414. [Google Scholar] [CrossRef]
- Matsuda, M.; Suizu, F.; Hirata, N.; Miyazaki, T.; Obuse, C.; Noguchi, M. Characterization of the interaction of influenza virus NS1 with Akt. Biochem. Biophys. Res. Commun. 2010, 395, 312–317. [Google Scholar] [CrossRef]
- Wang, X.; Shen, Y.; Qiu, Y.; Shi, Z.; Shao, D.; Chen, P.; Tong, G.; Ma, Z. The non-structural (NS1) protein of influenza A virus associates with p53 and inhibits p53-mediated transcriptional activity and apoptosis. Biochem. Biophys. Res. Commun. 2010, 395, 141–145. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, Y.; Zhou, X.; Yang, Z.; Liu, X.; Cao, Z.; Song, H.; He, Y.; Huang, P. The NS1 protein of influenza A virus interacts with heat shock protein Hsp90 in human alveolar basal epithelial cells: Implication for virus-induced apoptosis. Virol. J. 2011, 8, 181. [Google Scholar] [CrossRef]
- Han, X.Q.; Li, Z.H.; Chen, H.J.; Wang, H.Y.; Mei, L.; Wu, S.Q.; Zhang, T.Y.; Liu, B.H.; Lin, X.M. Influenza Virus A/Beijing/501/2009(H1N1) NS1 Interacts with beta-Tubulin and Induces Disruption of the Microtubule Network and Apoptosis on A549 Cells. PLoS ONE 2012, 7, e48340. [Google Scholar] [CrossRef]
- Burgui, I.; Aragon, T.; Ortin, J.; Nieto, A. PABP1 and eIF4GI associate with influenza virus NS1 protein in viral mRNA translation initiation complexes. J. Gen. Virol. 2003, 84, 3263–3274. [Google Scholar] [CrossRef]
- Lin, L.; Li, Y.; Pyo, H.M.; Lu, X.Y.; Raman, S.N.T.; Liu, Q.; Brown, E.G.; Zhou, Y. Identification of RNA Helicase A as a Cellular Factor That Interacts with Influenza A Virus NS1 Protein and Its Role in the Virus Life Cycle. J. Virol. 2012, 86, 1942–1954. [Google Scholar] [CrossRef]
- Chen, G.; Liu, C.H.; Zhou, L.; Krug, R.M. Cellular DDX21 RNA helicase inhibits influenza A virus replication but is counteracted by the viral NS1 protein. Cell Host Microbe. 2014, 15, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Garaigorta, U.; Falcon, A.M.; Ortin, J. Genetic analysis of influenza virus NS1 gene: A temperature-sensitive mutant shows defective formation of virus particles. J. Virol. 2005, 79, 15246–15257. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.; Hossain, M.J.; Hickman, D.; Perez, D.R.; Lamb, R.A. A new influenza virus virulence determinant: The NS1 protein four C-terminal residues modulate pathogenicity. Proc. Natl. Acad. Sci. USA 2008, 105, 4381–4386. [Google Scholar] [CrossRef]
- Seo, S.H.; Hoffmann, E.; Webster, R.G. Lethal H5N1 influenza viruses escape host anti-viral cytokine responses. Nat. Med. 2002, 8, 950–954. [Google Scholar] [CrossRef]
- Obenauer, J.C.; Denson, J.; Mehta, P.K.; Su, X.; Mukatira, S.; Finkelstein, D.B.; Xu, X.; Wang, J.; Ma, J.; Fan, Y.; et al. Large-scale sequence analysis of avian influenza isolates. Science 2006, 311, 1576–1580. [Google Scholar] [CrossRef] [PubMed]
- Soubies, S.M.; Volmer, C.; Croville, G.; Loupias, J.; Peralta, B.; Costes, P.; Lacroux, C.; Guerin, J.L.; Volmer, R. Species-specific contribution of the four C-terminal amino acids of influenza A virus NS1 protein to virulence. J. Virol. 2010, 84, 6733–6747. [Google Scholar] [CrossRef] [PubMed]
- Zielecki, F.; Semmler, I.; Kalthoff, D.; Voss, D.; Mauel, S.; Gruber, A.D.; Beer, M.; Wolff, T. Virulence determinants of avian H5N1 influenza A virus in mammalian and avian hosts: Role of the C-terminal ESEV motif in the viral NS1 protein. J. Virol. 2010, 84, 10708–10718. [Google Scholar] [CrossRef]
- Jiao, P.; Tian, G.; Li, Y.; Deng, G.; Jiang, Y.; Liu, C.; Liu, W.; Bu, Z.; Kawaoka, Y.; Chen, H. A single-amino-acid substitution in the NS1 protein changes the pathogenicity of H5N1 avian influenza viruses in mice. J. Virol. 2008, 82, 1146–1154. [Google Scholar] [CrossRef]
- Yu, J.; Li, X.; Wang, Y.; Li, B.; Li, H.; Li, Y.; Zhou, W.; Zhang, C.; Wang, Y.; Rao, Z.; et al. PDlim2 selectively interacts with the PDZ binding motif of highly pathogenic avian H5N1 influenza A virus NS1. PLoS ONE 2011, 6, e19511. [Google Scholar] [CrossRef]
- Liu, H.; Golebiewski, L.; Dow, E.C.; Krug, R.M.; Javier, R.T.; Rice, A.P. The ESEV PDZ-binding motif of the avian influenza A virus NS1 protein protects infected cells from apoptosis by directly targeting Scribble. J. Virol. 2010, 84, 11164–11174. [Google Scholar] [CrossRef]
- Thomas, M.; Kranjec, C.; Nagasaka, K.; Matlashewski, G.; Banks, L. Analysis of the PDZ binding specificities of Influenza A virus NS1 proteins. Virol. J. 2011, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, W.; Wang, G.; Su, Y.; Zhang, C.; Chen, X.; Xu, Y.; Li, K. The distinct binding properties between avian/human influenza A virus NS1 and Postsynaptic density protein-95 (PSD-95), and inhibition of nitric oxide production. Virol. J. 2011, 8, 298. [Google Scholar] [CrossRef] [PubMed]
- Golebiewski, L.; Liu, H.; Javier, R.T.; Rice, A.P. The avian influenza virus NS1 ESEV PDZ binding motif associates with Dlg1 and Scribble to disrupt cellular tight junctions. J. Virol. 2011, 85, 10639–10648. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Liu, H.; Rice, A.P. Regulation of interferon-beta by MAGI-1 and its interaction with influenza A virus NS1 protein with ESEV PBM. PLoS ONE 2012, 7, e41251. [Google Scholar] [CrossRef]
- Lamb, R.A.; Lai, C.J. Sequence of interrupted and uninterrupted mRNAs and cloned DNA coding for the two overlapping nonstructural proteins of influenza virus. Cell 1980, 21, 475–485. [Google Scholar] [CrossRef]
- Inglis, S.C.; Barrett, T.; Brown, C.M.; Almond, J.W. The smallest genome RNA segment of influenza virus contains two genes that may overlap. Proc. Natl. Acad. Sci. USA 1979, 76, 3790–3794. [Google Scholar] [CrossRef]
- Akarsu, H.; Burmeister, W.P.; Petosa, C.; Petit, I.; Muller, C.W.; Ruigrok, R.W.; Baudin, F. Crystal structure of the M1 protein-binding domain of the influenza A virus nuclear export protein (NEP/NS2). EMBO J. 2003, 22, 4646–4655. [Google Scholar] [CrossRef]
- Richardson, J.C.; Akkina, R.K. NS2 protein of influenza virus is found in purified virus and phosphorylated in infected cells. Arch. Virol. 1991, 116, 69–80. [Google Scholar] [CrossRef]
- Yasuda, J.; Nakada, S.; Kato, A.; Toyoda, T.; Ishihama, A. Molecular assembly of influenza virus: Association of the NS2 protein with virion matrix. Virology 1993, 196, 249–255. [Google Scholar] [CrossRef]
- Neumann, G.; Hughes, M.T.; Kawaoka, Y. Influenza A virus NS2 protein mediates vRNP nuclear export through NES-independent interaction with hCRM1. EMBO J. 2000, 19, 6751–6758. [Google Scholar] [CrossRef]
- Iwatsuki-Horimoto, K.; Horimoto, T.; Fujii, Y.; Kawaoka, Y. Generation of influenza A virus NS2 (NEP) mutants with an altered nuclear export signal sequence. J. Virol 2004, 78, 10149–10155. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Takizawa, N.; Watanabe, K.; Nagata, K.; Kobayashi, N. Crucial role of the influenza virus NS2 (NEP) C-terminal domain in M1 binding and nuclear export of vRNP. FEBS Lett. 2011, 585, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Boulo, S.; Akarsu, H.; Ruigrok, R.W.; Baudin, F. Nuclear traffic of influenza virus proteins and ribonucleoprotein complexes. Virus Res. 2007, 124, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Lommer, B.S.; Luo, M. Structural plasticity in influenza virus protein NS2 (NEP). J. Biol. Chem. 2002, 277, 7108–7117. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, R.E.; Talon, J.; Palese, P. The influenza virus NEP (NS2 protein) mediates the nuclear export of viral ribonucleoproteins. EMBO J. 1998, 17, 288–296. [Google Scholar] [CrossRef]
- Huang, S.; Chen, J.; Chen, Q.; Wang, H.; Yao, Y.; Chen, J.; Chen, Z. A second CRM1-dependent nuclear export signal in the influenza A virus NS2 protein contributes to the nuclear export of viral ribonucleoproteins. J. Virol. 2013, 87, 767–778. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, X.; Zhang, A.; Zhou, H.; Liu, Z.; Chen, H.; Jin, M. CHD3 facilitates vRNP nuclear export by interacting with NES1 of influenza A virus NS2. Cell Mol. Life Sci. 2015, 72, 971–982. [Google Scholar] [CrossRef]
- Pleschka, S.; Wolff, T.; Ehrhardt, C.; Hobom, G.; Planz, O.; Rapp, U.R.; Ludwig, S. Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat. Cell. Biol. 2001, 3, 301–305. [Google Scholar] [CrossRef]
- Gorai, T.; Goto, H.; Noda, T.; Watanabe, T.; Kozuka-Hata, H.; Oyama, M.; Takano, R.; Neumann, G.; Watanabe, S.; Kawaoka, Y. F1Fo-ATPase, F-type proton-translocating ATPase, at the plasma membrane is critical for efficient influenza virus budding. Proc. Natl. Acad. Sci. USA 2012, 109, 4615–4620. [Google Scholar] [CrossRef]
- Odagiri, T.; Tobita, K. Mutation in NS2, a nonstructural protein of influenza A virus, extragenically causes aberrant replication and expression of the PA gene and leads to generation of defective interfering particles. Proc. Natl. Acad. Sci. USA 1990, 87, 5988–5992. [Google Scholar] [CrossRef]
- Odagiri, T.; Tominaga, K.; Tobita, K.; Ohta, S. An amino acid change in the non-structural NS2 protein of an influenza A virus mutant is responsible for the generation of defective interfering (DI) particles by amplifying DI RNAs and suppressing complementary RNA synthesis. J. Gen. Virol. 1994, 75, 43–53. [Google Scholar] [CrossRef]
- Robb, N.C.; Smith, M.; Vreede, F.T.; Fodor, E. NS2/NEP protein regulates transcription and replication of the influenza virus RNA genome. J. Gen. Virol. 2009, 90, 1398–1407. [Google Scholar] [CrossRef] [PubMed]
- Bullido, R.; Gomez-Puertas, P.; Saiz, M.J.; Portela, A. Influenza A virus NEP (NS2 protein) downregulates RNA synthesis of model template RNAs. J. Virol. 2001, 75, 4912–4917. [Google Scholar] [CrossRef] [PubMed]
- Manz, B.; Brunotte, L.; Reuther, P.; Schwemmle, M. Adaptive mutations in NEP compensate for defective H5N1 RNA replication in cultured human cells. Nat. Commun. 2012, 3, 802. [Google Scholar] [CrossRef] [PubMed]
- Chua, M.A.; Schmid, S.; Perez, J.T.; Langlois, R.A.; Tenoever, B.R. Influenza A virus utilizes suboptimal splicing to coordinate the timing of infection. Cell Rep. 2013, 3, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Wu, J.; Liu, R.Y.; Li, J.; Song, L.; Teng, Y.; Sheng, C.; Liu, D.; Yao, C.; Chen, H.; et al. Interaction of NS2 with AIMP2 facilitates the switch from ubiquitination to SUMOylation of M1 in influenza A virus-infected cells. J. Virol. 2015, 89, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wang, L.; Li, S. Influenza A Virus-Host Protein Interactions Control Viral Pathogenesis. Int. J. Mol. Sci 2017, 18. [Google Scholar] [CrossRef]
- Chen, W.; Calvo, P.A.; Malide, D.; Gibbs, J.; Schubert, U.; Bacik, I.; Basta, S.; O’Neill, R.; Schickli, J.; Palese, P.; et al. A novel influenza A virus mitochondrial protein that induces cell death. Nat. Med. 2001, 7, 1306–1312. [Google Scholar] [CrossRef]
- Zamarin, D.; Garcia-Sastre, A.; Xiao, X.; Wang, R.; Palese, P. Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog. 2005, 1, e4. [Google Scholar] [CrossRef]
- Conenello, G.M.; Zamarin, D.; Perrone, L.A.; Tumpey, T.; Palese, P. A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog. 2007, 3, e141. [Google Scholar] [CrossRef]
- Chen, C.J.; Chen, G.W.; Wang, C.H.; Huang, C.H.; Wang, Y.C.; Shih, S.R. Differential localization and function of PB1-F2 derived from different strains of influenza A virus. J. Virol. 2010, 84, 10051–10062. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, J.S.; Malide, D.; Hornung, F.; Bennink, J.R.; Yewdell, J.W. The influenza A virus PB1-F2 protein targets the inner mitochondrial membrane via a predicted basic amphipathic helix that disrupts mitochondrial function. J. Virol. 2003, 77, 7214–7224. [Google Scholar] [CrossRef] [PubMed]
- Bruns, K.; Studtrucker, N.; Sharma, A.; Fossen, T.; Mitzner, D.; Eissmann, A.; Tessmer, U.; Roder, R.; Henklein, P.; Wray, V.; et al. Structural characterization and oligomerization of PB1-F2, a proapoptotic influenza A virus protein. J. Biol. Chem. 2007, 282, 353–363. [Google Scholar] [CrossRef]
- Yoshizumi, T.; Ichinohe, T.; Sasaki, O.; Otera, H.; Kawabata, S.; Mihara, K.; Koshiba, T. Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat. Commun. 2014, 5, 4713. [Google Scholar] [CrossRef]
- Chanturiya, A.N.; Basanez, G.; Schubert, U.; Henklein, P.; Yewdell, J.W.; Zimmerberg, J. PB1-F2, an influenza A virus-encoded proapoptotic mitochondrial protein, creates variably sized pores in planar lipid membranes. J. Virol. 2004, 78, 6304–6312. [Google Scholar] [CrossRef]
- McAuley, J.L.; Chipuk, J.E.; Boyd, K.L.; Van De Velde, N.; Green, D.R.; McCullers, J.A. PB1-F2 proteins from H5N1 and 20 century pandemic influenza viruses cause immunopathology. PLoS Pathog. 2010, 6, e1001014. [Google Scholar] [CrossRef]
- Jaworska, J.; Coulombe, F.; Downey, J.; Tzelepis, F.; Shalaby, K.; Tattoli, I.; Berube, J.; Rousseau, S.; Martin, J.G.; Girardin, S.E.; et al. NLRX1 prevents mitochondrial induced apoptosis and enhances macrophage antiviral immunity by interacting with influenza virus PB1-F2 protein. Proc. Natl. Acad. Sci. USA 2014, 111, E2110–E2119. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.T.; Ramos, I.; Hai, R.; Schmolke, M.; Garcia-Sastre, A.; Fernandez-Sesma, A.; Palese, P. The influenza virus protein PB1-F2 inhibits the induction of type I interferon at the level of the MAVS adaptor protein. PLoS Pathog. 2011, 7, e1002067. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.T.; Grant, A.; Manicassamy, B.; Palese, P. Influenza virus protein PB1-F2 inhibits the induction of type I interferon by binding to MAVS and decreasing mitochondrial membrane potential. J. Virol. 2012, 86, 8359–8366. [Google Scholar] [CrossRef]
- Reis, A.L.; McCauley, J.W. The influenza virus protein PB1-F2 interacts with IKKbeta and modulates NF-kappaB signalling. PLoS ONE 2013, 8, e63852. [Google Scholar] [CrossRef]
- Li, S.; Wang, L.; Berman, M.; Kong, Y.Y.; Dorf, M.E. Mapping a dynamic innate immunity protein interaction network regulating type I interferon production. Immunity 2011, 35, 426–440. [Google Scholar] [CrossRef] [PubMed]
- Leymarie, O.; Meyer, L.; Tafforeau, L.; Lotteau, V.; Costa, B.D.; Delmas, B.; Chevalier, C.; Le Goffic, R. Influenza virus protein PB1-F2 interacts with CALCOCO2 (NDP52) to modulate innate immune response. J. Gen. Virol. 2017, 98, 1196–1208. [Google Scholar] [CrossRef] [PubMed]
- Le Goffic, R.; Leymarie, O.; Chevalier, C.; Rebours, E.; Da Costa, B.; Vidic, J.; Descamps, D.; Sallenave, J.M.; Rauch, M.; Samson, M.; et al. Transcriptomic analysis of host immune and cell death responses associated with the influenza A virus PB1-F2 protein. PLoS Pathog. 2011, 7, e1002202. [Google Scholar] [CrossRef] [PubMed]
- Vidy, A.; Maisonnasse, P.; Da Costa, B.; Delmas, B.; Chevalier, C.; Le Goffic, R. The Influenza Virus Protein PB1-F2 Increases Viral Pathogenesis through Neutrophil Recruitment and NK Cells Inhibition. PLoS ONE 2016, 11, e0165361. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Monack, D.M. Molecular mechanisms of inflammasome activation during microbial infections. Immunol. Rev. 2011, 243, 174–190. [Google Scholar] [CrossRef] [PubMed]
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 2009, 30, 556–565. [Google Scholar] [CrossRef]
- Cheung, P.H.; Ye, Z.W.; Lee, T.T.; Chen, H.; Chan, C.P.; Jin, D.Y. PB1-F2 protein of highly pathogenic influenza A (H7N9) virus selectively suppresses RNA-induced NLRP3 inflammasome activation through inhibition of MAVS-NLRP3 interaction. J. Leukoc. Biol. 2020. [Google Scholar] [CrossRef]
- McAuley, J.L.; Tate, M.D.; MacKenzie-Kludas, C.J.; Pinar, A.; Zeng, W.; Stutz, A.; Latz, E.; Brown, L.E.; Mansell, A. Activation of the NLRP3 inflammasome by IAV virulence protein PB1-F2 contributes to severe pathophysiology and disease. PLoS Pathog. 2013, 9, e1003392. [Google Scholar] [CrossRef]
- Pinar, A.; Dowling, J.K.; Bitto, N.J.; Robertson, A.A.; Latz, E.; Stewart, C.R.; Drummond, G.R.; Cooper, M.A.; McAuley, J.L.; Tate, M.D.; et al. PB1-F2 Peptide Derived from Avian Influenza A Virus H7N9 Induces Inflammation via Activation of the NLRP3 Inflammasome. J. Biol. Chem. 2017, 292, 826–836. [Google Scholar] [CrossRef]
- Yabal, M.; Calleja, D.J.; Simpson, D.S.; Lawlor, K.E. Stressing out the mitochondria: Mechanistic insights into NLRP3 inflammasome activation. J. Leukoc. Biol. 2019, 105, 377–399. [Google Scholar] [CrossRef]
- Mazur, I.; Anhlan, D.; Mitzner, D.; Wixler, L.; Schubert, U.; Ludwig, S. The proapoptotic influenza A virus protein PB1-F2 regulates viral polymerase activity by interaction with the PB1 protein. Cell Microbiol. 2008, 10, 1140–1152. [Google Scholar] [CrossRef] [PubMed]
- Hai, R.; Schmolke, M.; Varga, Z.T.; Manicassamy, B.; Wang, T.T.; Belser, J.A.; Pearce, M.B.; Garcia-Sastre, A.; Tumpey, T.M.; Palese, P. PB1-F2 expression by the 2009 pandemic H1N1 influenza virus has minimal impact on virulence in animal models. J. Virol. 2010, 84, 4442–4450. [Google Scholar] [CrossRef] [PubMed]
- McAuley, J.L.; Zhang, K.; McCullers, J.A. The effects of influenza A virus PB1-F2 protein on polymerase activity are strain specific and do not impact pathogenesis. J. Virol. 2010, 84, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Conenello, G.M.; Tisoncik, J.R.; Rosenzweig, E.; Varga, Z.T.; Palese, P.; Katze, M.G. A single N66S mutation in the PB1-F2 protein of influenza A virus increases virulence by inhibiting the early interferon response in vivo. J. Virol. 2011, 85, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Schmolke, M.; Manicassamy, B.; Pena, L.; Sutton, T.; Hai, R.; Varga, Z.T.; Hale, B.G.; Steel, J.; Perez, D.R.; Garcia-Sastre, A. Differential contribution of PB1-F2 to the virulence of highly pathogenic H5N1 influenza A virus in mammalian and avian species. PLoS Pathog. 2011, 7, e1002186. [Google Scholar] [CrossRef]
- Lee, J.; Henningson, J.; Ma, J.; Duff, M.; Lang, Y.; Li, Y.; Li, Y.; Nagy, A.; Sunwoo, S.; Bawa, B.; et al. Effects of PB1-F2 on the pathogenicity of H1N1 swine influenza virus in mice and pigs. J. Gen. Virol. 2017, 98, 31–42. [Google Scholar] [CrossRef]
- Alymova, I.V.; Green, A.M.; van de Velde, N.; McAuley, J.L.; Boyd, K.L.; Ghoneim, H.E.; McCullers, J.A. Immunopathogenic and antibacterial effects of H3N2 influenza A virus PB1-F2 map to amino acid residues 62, 75, 79, and 82. J. Virol. 2011, 85, 12324–12333. [Google Scholar] [CrossRef]
- Weeks-Gorospe, J.N.; Hurtig, H.R.; Iverson, A.R.; Schuneman, M.J.; Webby, R.J.; McCullers, J.A.; Huber, V.C. Naturally occurring swine influenza A virus PB1-F2 phenotypes that contribute to superinfection with Gram-positive respiratory pathogens. J. Virol. 2012, 86, 9035–9043. [Google Scholar] [CrossRef]
- Alymova, I.V.; Samarasinghe, A.; Vogel, P.; Green, A.M.; Weinlich, R.; McCullers, J.A. A novel cytotoxic sequence contributes to influenza A viral protein PB1-F2 pathogenicity and predisposition to secondary bacterial infection. J. Virol. 2014, 88, 503–515. [Google Scholar] [CrossRef]
- Cheng, Y.Y.; Yang, S.R.; Wang, Y.T.; Lin, Y.H.; Chen, C.J. Amino Acid Residues 68-71 Contribute to Influenza A Virus PB1-F2 Protein Stability and Functions. Front. Microbiol. 2017, 8, 692. [Google Scholar] [CrossRef]
- Firth, A.E.; Jagger, B.W.; Wise, H.M.; Nelson, C.C.; Parsawar, K.; Wills, N.M.; Napthine, S.; Taubenberger, J.K.; Digard, P.; Atkins, J.F. Ribosomal frameshifting used in influenza A virus expression occurs within the sequence UCC_UUU_CGU and is in the +1 direction. Open Biol. 2012, 2, 120109. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Jagger, B.W.; Wise, H.M.; Digard, P.; Holmes, E.C.; Taubenberger, J.K. Evolutionary conservation of the PA-X open reading frame in segment 3 of influenza A virus. J. Virol. 2012, 86, 12411–12413. [Google Scholar] [CrossRef] [PubMed]
- Desmet, E.A.; Bussey, K.A.; Stone, R.; Takimoto, T. Identification of the N-terminal domain of the influenza virus PA responsible for the suppression of host protein synthesis. J. Virol. 2013, 87, 3108–3118. [Google Scholar] [CrossRef] [PubMed]
- Khaperskyy, D.A.; Schmaling, S.; Larkins-Ford, J.; McCormick, C.; Gaglia, M.M. Selective Degradation of Host RNA Polymerase II Transcripts by Influenza A Virus PA-X Host Shutoff Protein. PLoS Pathog 2016, 12, e1005427. [Google Scholar] [CrossRef] [PubMed]
- Khaperskyy, D.A.; McCormick, C. Timing Is Everything: Coordinated Control of Host Shutoff by Influenza A Virus NS1 and PA-X Proteins. J. Virol. 2015, 89, 6528–6531. [Google Scholar] [CrossRef] [PubMed]
- Gaucherand, L.; Porter, B.K.; Levene, R.E.; Price, E.L.; Schmaling, S.K.; Rycroft, C.H.; Kevorkian, Y.; McCormick, C.; Khaperskyy, D.A.; Gaglia, M.M. The Influenza A Virus Endoribonuclease PA-X Usurps Host mRNA Processing Machinery to Limit Host Gene Expression. Cell Rep. 2019, 27, 776–792.e7. [Google Scholar] [CrossRef]
- Hayashi, T.; Chaimayo, C.; McGuinness, J.; Takimoto, T. Critical Role of the PA-X C-Terminal Domain of Influenza A Virus in Its Subcellular Localization and Shutoff Activity. J. Virol. 2016, 90, 7131–7141. [Google Scholar] [CrossRef]
- Lee, J.; Yu, H.; Li, Y.; Ma, J.; Lang, Y.; Duff, M.; Henningson, J.; Liu, Q.; Li, Y.; Nagy, A.; et al. Impacts of different expressions of PA-X protein on 2009 pandemic H1N1 virus replication, pathogenicity and host immune responses. Virology 2017, 504, 25–35. [Google Scholar] [CrossRef]
- Gao, H.; Sun, H.; Hu, J.; Qi, L.; Wang, J.; Xiong, X.; Wang, Y.; He, Q.; Lin, Y.; Kong, W.; et al. Twenty amino acids at the C-terminus of PA-X are associated with increased influenza A virus replication and pathogenicity. J. Gen. Virol. 2015, 96, 2036–2049. [Google Scholar] [CrossRef]
- Oishi, K.; Yamayoshi, S.; Kawaoka, Y. Identification of novel amino acid residues of influenza virus PA-X that are important for PA-X shutoff activity by using yeast. Virology 2018, 516, 71–75. [Google Scholar] [CrossRef]
- Oishi, K.; Yamayoshi, S.; Kozuka-Hata, H.; Oyama, M.; Kawaoka, Y. N-Terminal Acetylation by NatB Is Required for the Shutoff Activity of Influenza A Virus PA-X. Cell Rep. 2018, 24, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; MacDonald, L.A.; Takimoto, T. Influenza A Virus Protein PA-X Contributes to Viral Growth and Suppression of the Host Antiviral and Immune Responses. J. Virol. 2015, 89, 6442–6452. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Sun, Y.; Hu, J.; Qi, L.; Wang, J.; Xiong, X.; Wang, Y.; He, Q.; Lin, Y.; Kong, W.; et al. The contribution of PA-X to the virulence of pandemic 2009 H1N1 and highly pathogenic H5N1 avian influenza viruses. Sci. Rep. 2015, 5, 8262. [Google Scholar] [CrossRef] [PubMed]
- Rigby, R.E.; Wise, H.M.; Smith, N.; Digard, P.; Rehwinkel, J. PA-X antagonises MAVS-dependent accumulation of early type I interferon messenger RNAs during influenza A virus infection. Sci. Rep. 2019, 9, 7216. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Mo, Y.; Wang, X.; Gu, M.; Hu, Z.; Zhong, L.; Wu, Q.; Hao, X.; Hu, S.; Liu, W.; et al. PA-X decreases the pathogenicity of highly pathogenic H5N1 influenza A virus in avian species by inhibiting virus replication and host response. J. Virol. 2015, 89, 4126–4142. [Google Scholar] [CrossRef]
- Gong, X.Q.; Sun, Y.F.; Ruan, B.Y.; Liu, X.M.; Wang, Q.; Yang, H.M.; Wang, S.Y.; Zhang, P.; Wang, X.H.; Shan, T.L.; et al. PA-X protein decreases replication and pathogenicity of swine influenza virus in cultured cells and mouse models. Vet. Microbiol. 2017, 205, 66–70. [Google Scholar] [CrossRef]
- Gao, H.; Xu, G.; Sun, Y.; Qi, L.; Wang, J.; Kong, W.; Sun, H.; Pu, J.; Chang, K.C.; Liu, J. PA-X is a virulence factor in avian H9N2 influenza virus. J. Gen. Virol. 2015, 96, 2587–2594. [Google Scholar] [CrossRef]
- Tauber, S.; Ligertwood, Y.; Quigg-Nicol, M.; Dutia, B.M.; Elliott, R.M. Behaviour of influenza A viruses differentially expressing segment 2 gene products in vitro and in vivo. J. Gen. Virol. 2012, 93, 840–849. [Google Scholar] [CrossRef]
- Konig, R.; Stertz, S.; Zhou, Y.; Inoue, A.; Hoffmann, H.H.; Bhattacharyya, S.; Alamares, J.G.; Tscherne, D.M.; Ortigoza, M.B.; Liang, Y.; et al. Human host factors required for influenza virus replication. Nature 2010, 463, 813–817. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, R.; Li, Q.; Wang, F.; Zhu, B.; Zheng, M.; Cui, H.; Wen, J.; Zhao, G. Host cell interactome of PB1 N40 protein of H5N1 influenza A virus in chicken cells. J. Proteom. 2019, 197, 34–41. [Google Scholar] [CrossRef]
- Clifford, M.; Twigg, J.; Upton, C. Evidence for a novel gene associated with human influenza A viruses. Virol. J. 2009, 6, 198. [Google Scholar] [CrossRef] [PubMed]
- Zhirnov, O.P.; Poyarkov, S.V.; Vorob’eva, I.V.; Safonova, O.A.; Malyshev, N.A.; Klenk, H.D. Segment NS of influenza A virus contains an additional gene NSP in positive-sense orientation. Dokl. Biochem. Biophys. 2007, 414, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.N.; Chen, G.W.; Chen, C.J.; Kuo, R.L.; Shih, S.R. Computational analysis and mapping of novel open reading frames in influenza A viruses. PLoS ONE 2014, 9, e115016. [Google Scholar] [CrossRef]
- Pica, N.; Langlois, R.A.; Krammer, F.; Margine, I.; Palese, P. NS1-truncated live attenuated virus vaccine provides robust protection to aged mice from viral challenge. J. Virol. 2012, 86, 10293–10301. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.N.; Langley, W.A.; Carnero, E.; Garcia-Sastre, A.; Ahmed, R. Immunization with Live Attenuated Influenza Viruses That Express Altered NS1 Proteins Results in Potent and Protective Memory CD8(+) T-Cell Responses. J. Virol. 2010, 84, 1847–1855. [Google Scholar] [CrossRef] [PubMed]
- Mossler, C.; Groiss, F.; Wolzt, M.; Wolschek, M.; Seipelt, J.; Muster, T. Phase I/II trial of a replication-deficient trivalent influenza virus vaccine lacking NS1. Vaccine 2013, 31, 6194–6200. [Google Scholar] [CrossRef]
- Meyer, L.; Sausset, A.; Sedano, L.; Da Costa, B.; Le Goffic, R.; Delmas, B. Codon Deletions in the Influenza A Virus PA Gene Generate Temperature-Sensitive Viruses. J. Virol. 2016, 90, 3684–3693. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, W.; Wang, L.; Li, S. Roles of the Non-Structural Proteins of Influenza A Virus. Pathogens 2020, 9, 812. https://doi.org/10.3390/pathogens9100812
Hao W, Wang L, Li S. Roles of the Non-Structural Proteins of Influenza A Virus. Pathogens. 2020; 9(10):812. https://doi.org/10.3390/pathogens9100812
Chicago/Turabian StyleHao, Wenzhuo, Lingyan Wang, and Shitao Li. 2020. "Roles of the Non-Structural Proteins of Influenza A Virus" Pathogens 9, no. 10: 812. https://doi.org/10.3390/pathogens9100812
APA StyleHao, W., Wang, L., & Li, S. (2020). Roles of the Non-Structural Proteins of Influenza A Virus. Pathogens, 9(10), 812. https://doi.org/10.3390/pathogens9100812