High Adhesion and Increased Cell Death Contribute to Strong Biofilm Formation in Klebsiella pneumoniae


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
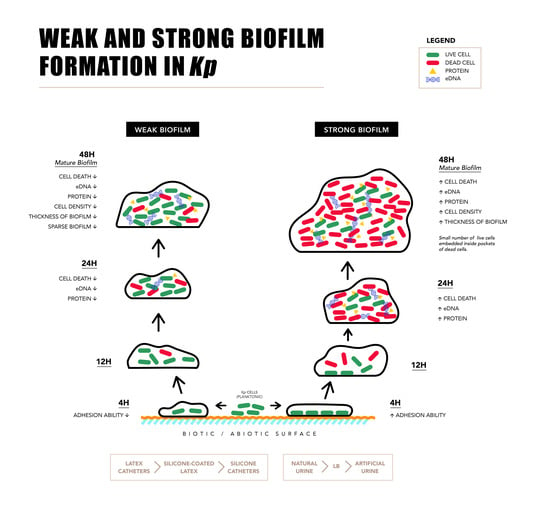
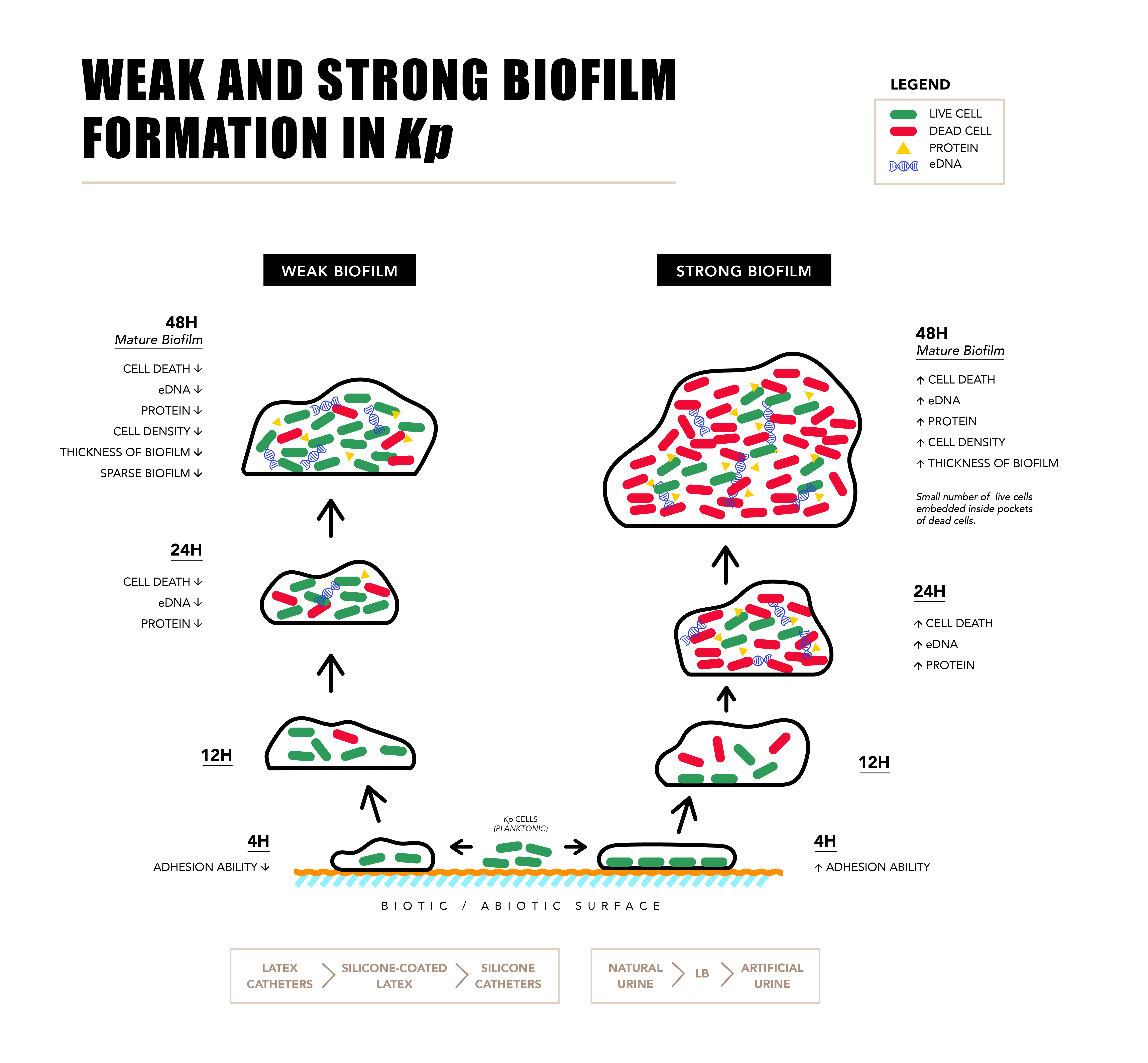
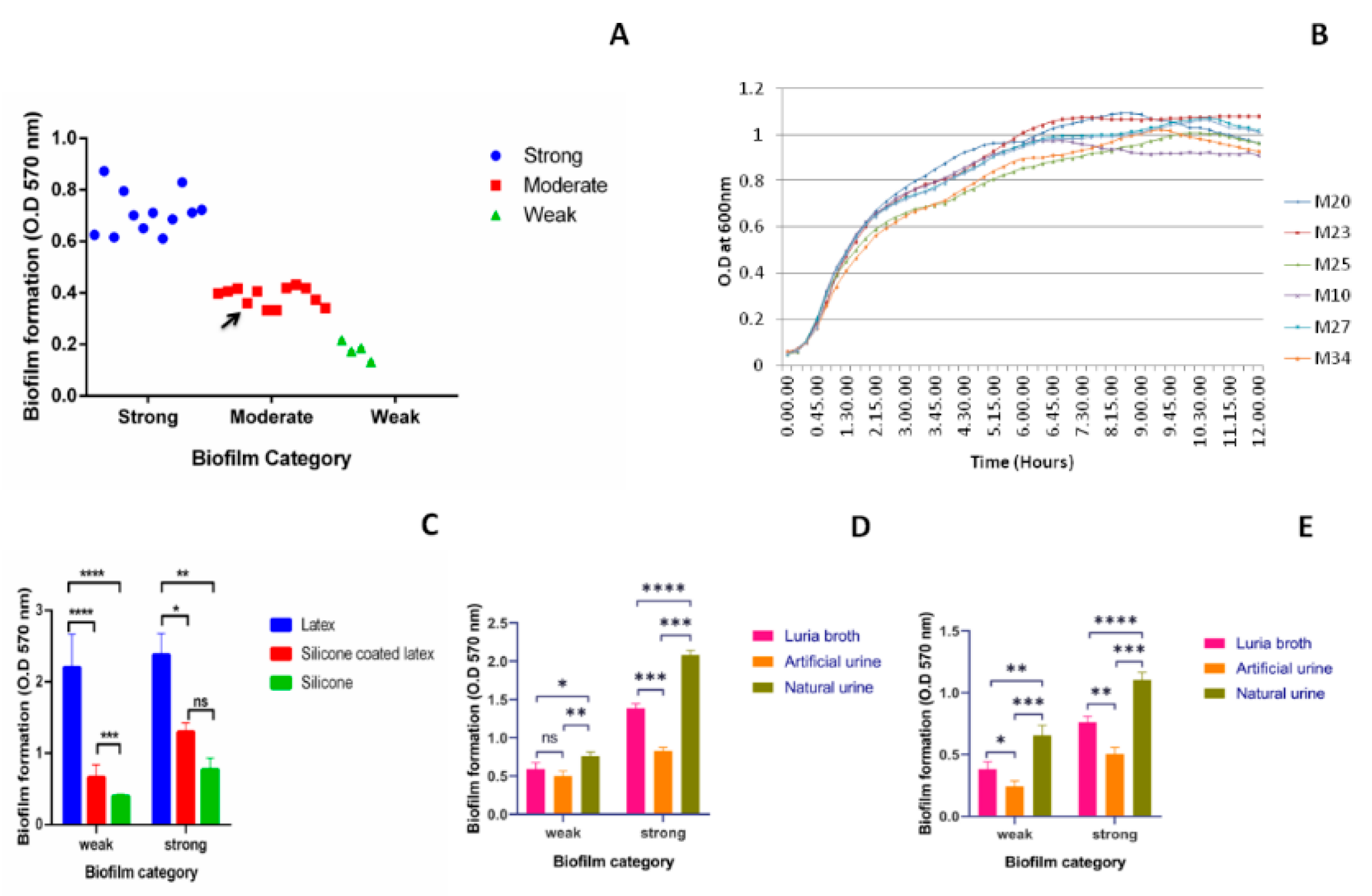
2.1. Biofilm Formation by Uropathogenic Kp
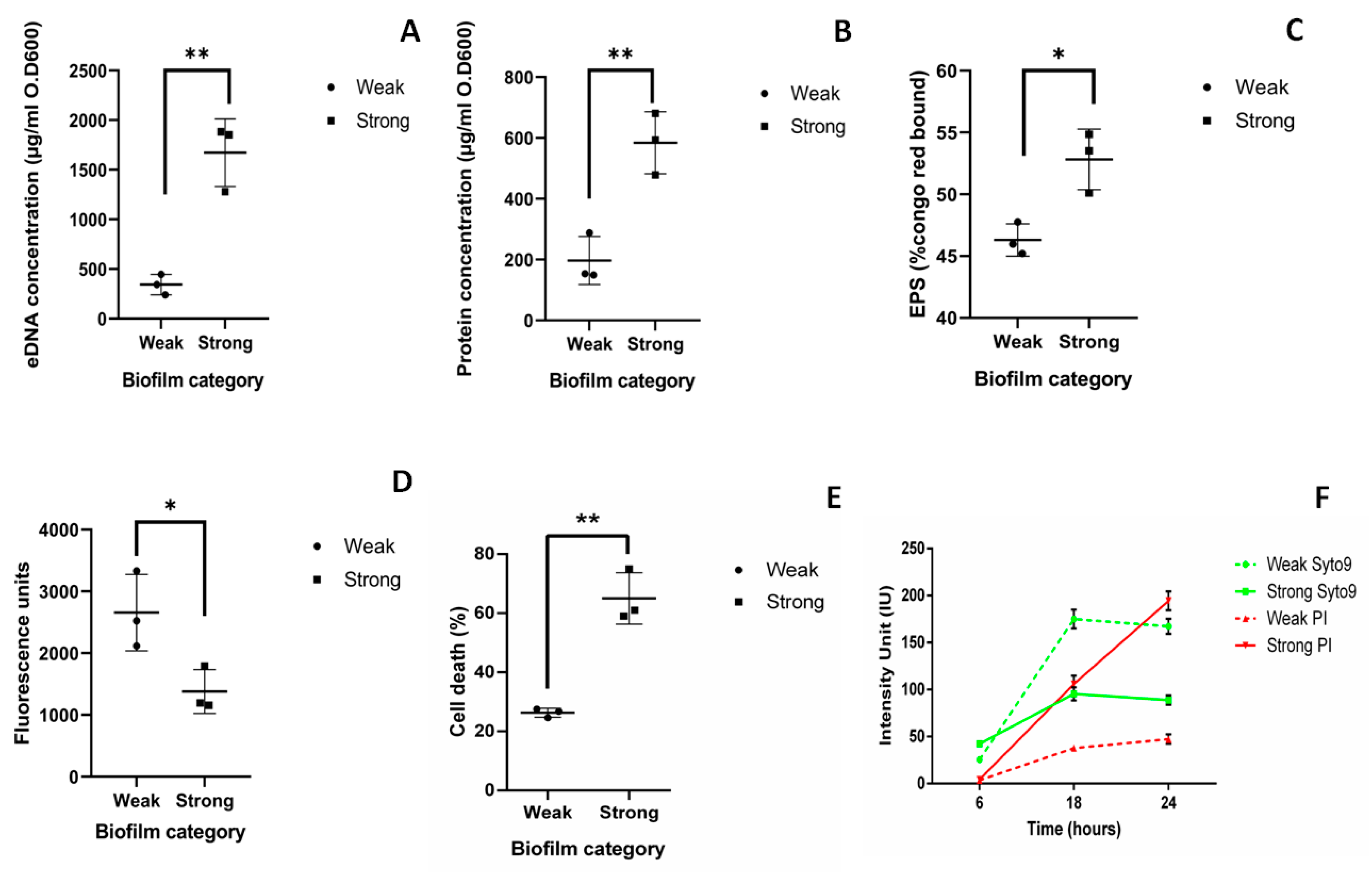
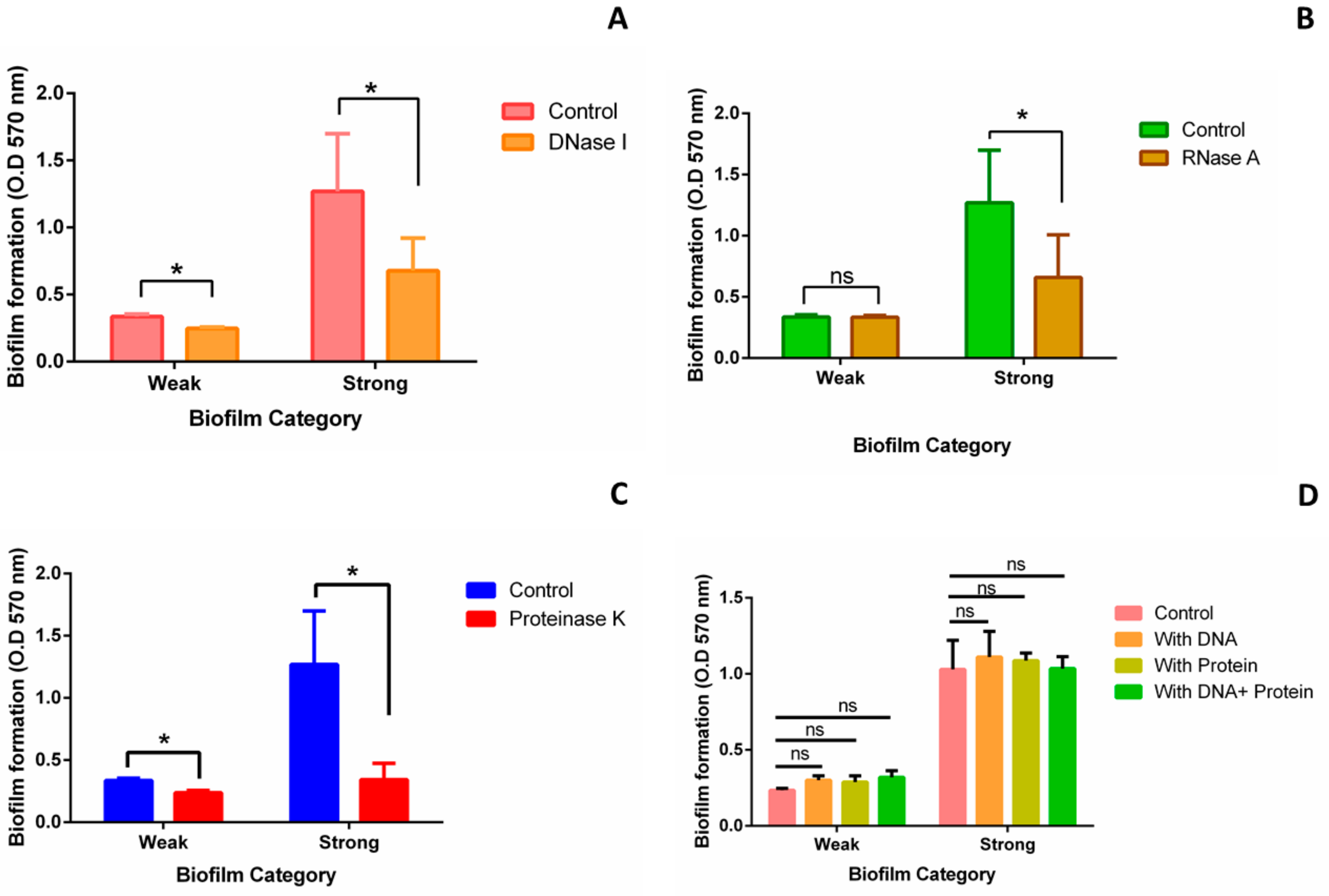
2.2. Components of Strong and Weak Biofilm Matrix
Inhibition and Addition Assay
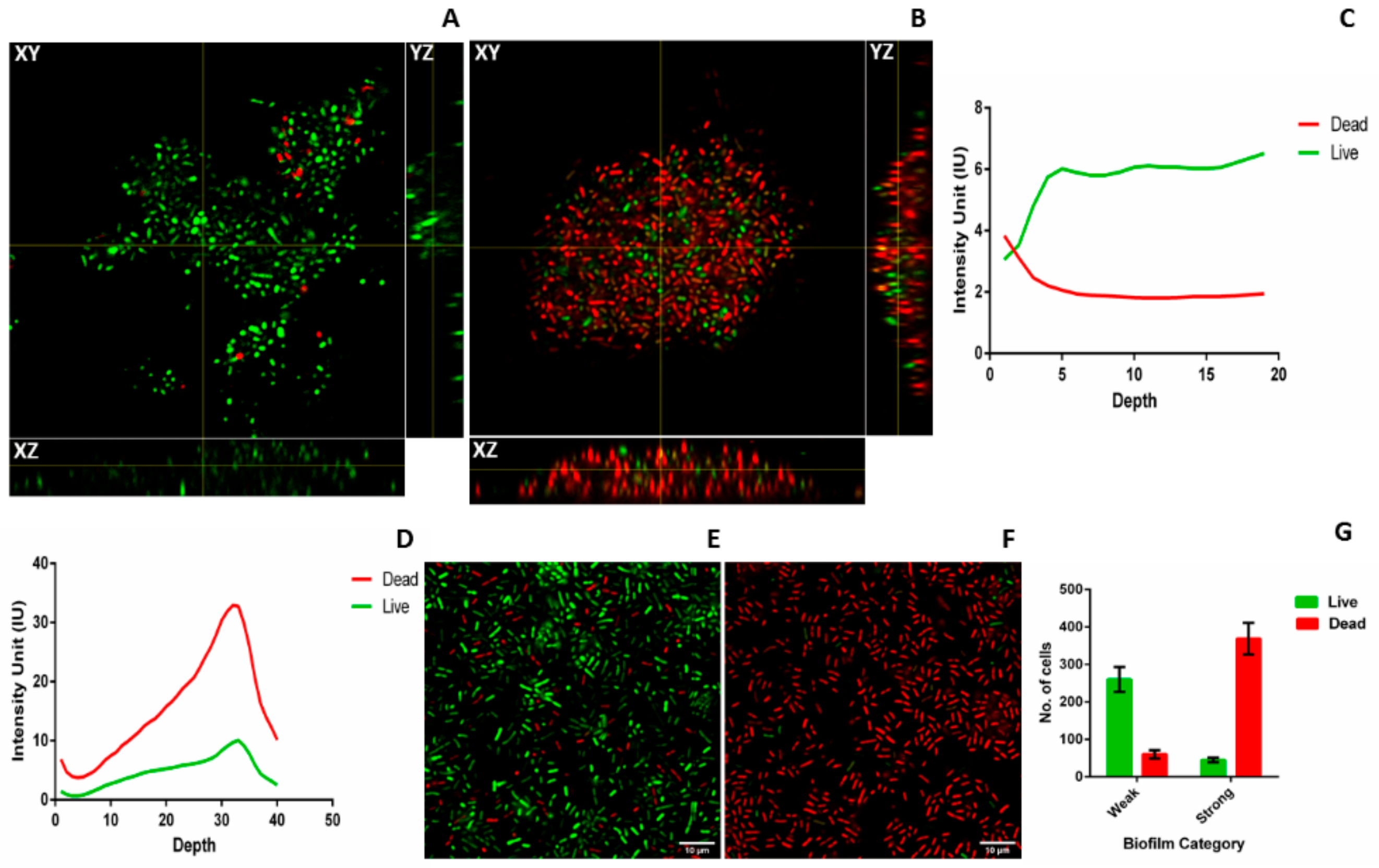
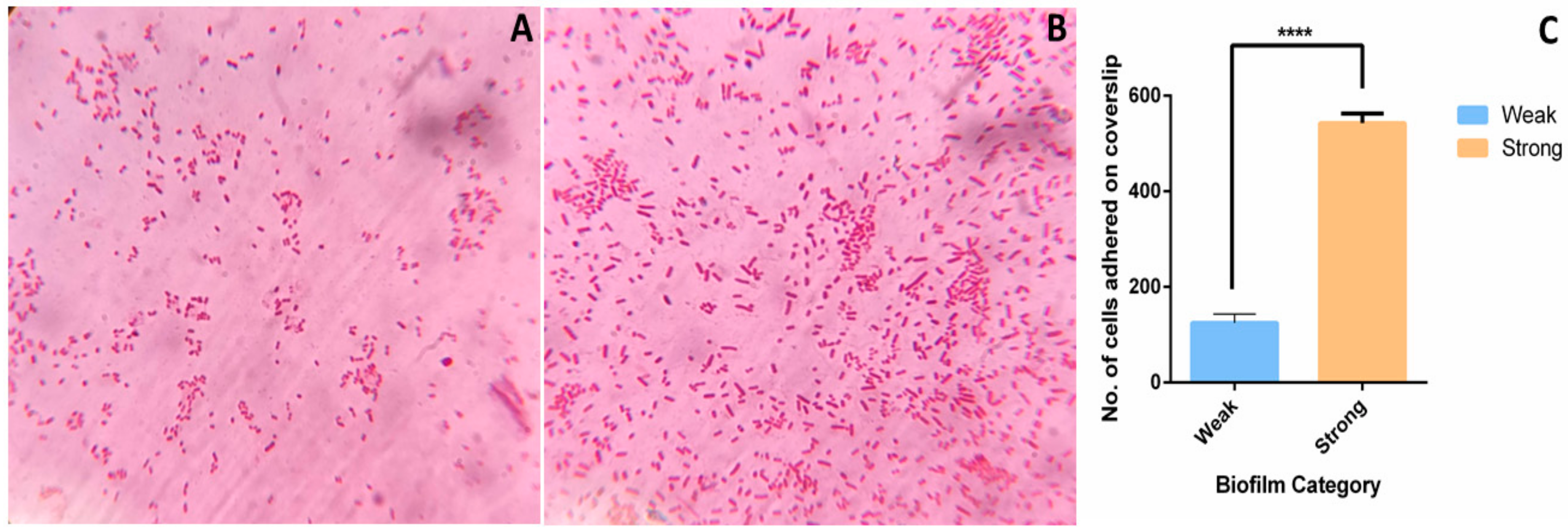
2.3. Microscopy of Weak and Strong Biofilm
3. Discussion
4. Materials and Methods
4.1. Bacterial Isolates and Growth Conditions
4.2. Quantification of Biofilm Formation
4.3. Growth Curve
4.4. Quantification of Biofilm on Catheters
4.5. Quantification of Components of Biofilm Matrix
4.5.1. eDNA Quantification
4.5.2. Extracellular Protein Quantification
4.5.3. Exopolysaccharide (EPS) Quantification
4.5.4. Live Dead Assay
4.6. Resazurin Assay
4.7. Time Bound Live Dead Assay
4.8. Inhibition Assays
4.9. Addition Assay
4.10. Microscopy of Strong and Weak Biofilms
4.10.1. CLSM
4.10.2. FEG-SEM
4.10.3. Cell Adhesion Assay
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Podschun, R.; Ullmann, U. Klebsiella spp. as nosocomial pathogens: Epidemiology, taxonomy, typing methods, and pathogenicity factors. Clin. Microbiol. Rev. 1998, 11, 589–603. [Google Scholar] [CrossRef] [PubMed]
- Niveditha, S.; Pramodhini, S.; Umadevi, S.; Kumar, S.; Stephen, S. The isolation and the biofilm formation of uropathogens in the patients with catheter associated urinary tract infections (UTIs). J. Clin. Diagn. Res 2012, 6, 1478–1482. [Google Scholar] [CrossRef] [PubMed]
- Høiby, N.; Ciofu, O.; Johansen, H.K.; Song, Z.J.; Moser, C.; Jensen, P.Ø.; Molin, S.; Givskov, M.; Tolker-Nielsen, T.; Bjarnsholt, T. The clinical impact of bacterial biofilms. Int. J. Oral. Sci. 2011, 3, 55–65. [Google Scholar] [CrossRef]
- Geraci, D.M.; Bonura, C.; Giuffrè, M.; Saporito, L.; Graziano, G.; Aleo, A.; Fasciana, T.; Di Bernardo, F.; Stampone, T.; Palma, D.M.; et al. Is the monoclonal spread of the ST258, KPC-3-producing clone being replaced in southern Italy by the dissemination of multiple clones of carbapenemnonsusceptible, KPC-3-producing Klebsiella pneumoniae? Clin. Microbiol. Infect. 2015, 21, 15–17. [Google Scholar] [CrossRef] [PubMed]
- Bonura, C.; Giuffrè, M.; Aleo, A.; Fasciana, T.; Di Bernardo, F.; Stampone, T.; Giammanco, A.; MDR-GNWorking Group Palma, D.M.; Mammina, C. An Update of the Evolving Epidemic of blaKPC Carrying Klebsiella pneumoniae in Sicily, Italy, 2014: Emergence of Multiple Non-ST258 Clones. PLoS ONE 2015, 10, e0132936. [Google Scholar] [CrossRef] [PubMed]
- Nicolle, L.E. Catheter associated urinary tract infections. Antimicrob. Resist. Infect. Control. 2014, 3, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.L.Y.; Ulett, G.C.; Mabbett, A.N.; Beatson, S.A.; Webb, R.I.; Monaghan, W.; Nimmo, G.R.; Looke, D.F.; McEwan, A.G.; Schembri, M.A. Identification of type-3 fimbriae in uro-pathogenic Escherichia coli reveals a role in biofilm formation. J. Bacteriol. 2008, 190, 1054–1063. [Google Scholar] [CrossRef]
- Bandeira, M.; Carvalho, P.A.; Duarte, A.; Jordao, L. Exploring Dangerous Connections between Klebsiella pneumoniae Biofilms and Healthcare-Associated Infections. Pathogens 2014, 3, 720–731. [Google Scholar] [CrossRef]
- Lewis, K. Multidrug tolerance of biofilms and persister cells. Bacterial Biofilms 2008, 322, 107–131. [Google Scholar]
- Calà, C.; Amodio, E.; Di Carlo, E.; Virruso, R.; Fasciana, T.; Giammanco, A. Biofilm production in Staphylococcus epidermidis strains, isolated from the skin of hospitalized patients: genetic and phenotypic characteristics. New. Microbiol. 2015, 38, 521–529. [Google Scholar]
- Vuotto, C.; Longo, F.; Pascolini, C.; Donelli, G.; Balice, M.P.; Libori, M.F.; Tiracchia, V.; Salvia, A.; Varaldo, P.E. Biofilm formation and antibiotic resistance in Klebsiella pneumoniae urinary strains. J. Appl. Microbiol. 2017, 123, 1003–1018. [Google Scholar] [CrossRef] [PubMed]
- Ostria-Hernandez, M.L.; Juárez-de la Rosa, K.C.; Arzate-Barbosa, P.; Lara-Hernández, A.; Sakai, F.; Ibarra, J.A.; Castro-Escarpulli, G.; Vidal, J.E. Nosocomial, Multidrug-Resistant Klebsiella pneumoniae Strains Isolated from Mexico City Produce Robust Biofilms on Abiotic Surfaces but Not on Human Lung Cells. Microb. Drug. Resist. 2018, 24, 422–433. [Google Scholar] [CrossRef] [PubMed]
- De Campos, P.A.; Royer, S.; da Fonseca Batistao, D.W.; Araújo, B.F.; Queiroz, L.L.; de Brito, C.S.; Gontijo-Filho, P.P.; Ribas, R.M. Multidrug resistance related to biofilm formation in Acinetobacter baumannii and Klebsiella pneumoniae clinical strains from different pulsotypes. Curr. Microbiol. 2016, 72, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Khodadadian, R.; Rahdar, H.A.; Javadi, A.; Safari, M.; Khorshidi, A. Detection of VIM-1 and IMP-1 genes in Klebsiella pneumoniae and relationship with biofilm formation. Microb. Pathog. 2018, 115, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Schroll, C.; Barken, K.B.; Krogfelt, K.A.; Struve, C. Role of type 1 and type 3 fimbriae in Klebsiella pneumoniae biofilm formation. BMC Microbiol. 2010, 10, 179–188. [Google Scholar] [CrossRef]
- Stahlhut, S.G.; Struve, C.; Krogfelt, K.A.; Reisner, A. Biofilm formation of Klebsiella pneumoniae on urethral catheters requires either type 1 or type 3 fimbriae. FEMS Immunol. Med. Microbiol. 2012, 65, 350–359. [Google Scholar] [CrossRef]
- Peeters, E.; Nelis, H.J.; Coenye, T. Comparison of multiple methods for quantification of microbial biofilms grown in microtiter plates. J. Microbiol. Methods 2008, 72, 157–165. [Google Scholar] [CrossRef]
- Diago-Navarro, E.; Chen, L.; Passet, V.; Burack, S.; Ulacia-Hernando, A.; Kodiyanplakkal, R.P.; Levi, M.H.; Brisse, S.; Kreiswirth, B.N.; Fries, B.C. Carbapenem-resistant Klebsiella pneumoniae exhibit variability in capsular polysaccharide and capsule associated virulence traits. J. Infect. Dis. 2014, 210, 803–813. [Google Scholar] [CrossRef]
- NicolauKorres, A.M.; Aquije, G.M.D.F.V.; Buss, D.S.; Ventura, J.A.; Fernandes, P.M.B.; Fernandes, A.A.R. Comparison of biofilm and attachment mechanisms of a phytopathological and clinical isolate of Klebsiella pneumonia subsp. pneumoniae. Sci. World J. 2013, 2013, 925375. [Google Scholar] [CrossRef]
- Revdiwala, S.; Rajdev, B.M.; Mulla, S. Characterization of bacterial etiologic agents of biofilm formation in medical devices in critical care setup. Crit. Care Res. Pract. 2012, 2012. [Google Scholar] [CrossRef]
- Anderl, J.N.; Zahller, J.; Roe, F.; Stewart, P.S. Role of nutrient limitation and stationary-phase existence in Klebsiella pneumoniae biofilm resistance to ampicillin and ciprofloxacin. Antimicrob. Agents Chemother. 2003, 47, 251–1256. [Google Scholar] [CrossRef]
- Zahller, J.; Stewart, P.S. Transmission electron microscopic study of antibiotic action on Klebsiella pneumoniae biofilm. Antimicrob. Agents Chemother. 2002, 46, 2679–2683. [Google Scholar] [CrossRef]
- Crémet, L.; Corvec, S.; Batard, E.; Auger, M.; Lopez, I.; Pagniez, F.; Dauvergne, S.; Caroff, N. Comparison of three methods to study biofilm formation by clinical strains of Escherichia coli. Diagn. Microbiol. Infect. Dis. 2013, 75, 252–255. [Google Scholar] [CrossRef]
- Balestrino, D.; Ghigo, J.M.; Charbonnel, N.; Haagensen, J.A.; Forestier, C. The characterization of functions involved in the establishment and maturation of Klebsiella pneumoniae in vitro biofilm reveals dual roles for surface exopolysaccharides. Environ. Microbiol. 2008, 10, 685–701. [Google Scholar] [CrossRef]
- Feneley, R.C.; Hopley, I.B.; Wells, P.N. Urinary catheters: History, current status, adverse events and research agenda. J. Med. Eng. Technol 2015, 39, 459–470. [Google Scholar] [CrossRef]
- Donlan, R.M. Biofilms and device-associated infections. Emerg. Infect. Dis. 2001, 7, 277–281. [Google Scholar] [CrossRef]
- Tunney, M.M.; Jones, D.S.; Gorman, S.P. Biofilm and biofilm-related encrustation of urinary tract devices. Methods Enzymol. 1999, 310, 558–566. [Google Scholar]
- Lee, K.H.; Park, S.J.; Choi, S.; Uh, Y.; Park, J.Y.; Han, K.H. The influence of urinary catheter materials on forming biofilms of microorganisms. J. Bacteriol. Virol. 2017, 47, 32–40. [Google Scholar] [CrossRef]
- Bandeira, M.; Borges, V.; Gomes, J.P.; Duarte, A.; Jordao, L. Insights on Klebsiella pneumoniae biofilms assembled on Different Surfaces using phenotypic and genotypic approaches. Microorganisms 2017, 5, 16. [Google Scholar] [CrossRef]
- Hu, W.; Li, L.; Sharma, S.; Wang, J.; McHardy, I.; Lux, R.; Yang, Z.; He, X.; Gimzewski, J.K.; Li, Y.; et al. DNA builds and strengthens the extracellular matrix in Myxococcus xanthus biofilms by interacting with exopolysaccharides. PLoS ONE 2012, 7, e51905. [Google Scholar] [CrossRef]
- Huseby, M.J.; Kruse, A.C.; Digre, J.; Kohler, P.L.; Vocke, J.A.; Mann, E.E.; Bayles, K.W.; Bohach, G.A.; Schlievert, P.M.; Ohlendorf, D.H.; et al. Beta toxin catalyzes formation of nucleoprotein matrix in staphylococcal biofilms. Proc. Natl. Acad. Sci. USA 2010, 107, 14407–14412. [Google Scholar] [CrossRef]
- Domenech, M.; García, E.; Prieto, A.; Moscoso, M. Insight into the composition of the intercellular matrix of Streptococcus pneumoniae biofilms. Environ. Microbiol. 2013, 15, 502–516. [Google Scholar] [CrossRef]
- Okshevsky, M.; Meyer, R.L. The role of extracellular DNA in the establishment, maintenance and perpetuation of bacterial biofilms. Crit. Rev. Microbiol. 2015, 41, 341–352. [Google Scholar] [CrossRef]
- Flemming, H.C.; Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. [Google Scholar] [CrossRef]
- Jakubovics, N.S.; Shields, R.C.; Rajarajan, N.; Burgess, J.G. Life after death: The critical role of extracellular DNA in microbial biofilms. Lett. Appl Microbiol. 2013, 57, 467–475. [Google Scholar] [CrossRef]
- Tetz, G.V.; Artemenko, N.K.; Tetz, V.V. Effect of DNase and antibiotics on biofilm characteristics. Antimicrob. Agents Chemother. 2009, 53, 1204–1209. [Google Scholar] [CrossRef]
- Harmsen, M.; Lappann, M.; Knøchel, S.; Molin, S. Role of extracellular DNA during biofilm formation by listeria monocytogenes. Appl. Environ. Microbiol. 2010, 76, 2271–2279. [Google Scholar] [CrossRef]
- Bayles, K.W. The biological role of death and lysis in biofilm development. Nat. Rev. Microbiol. 2007, 5, 721–726. [Google Scholar] [CrossRef]
- Lewis, K. Programmed death in bacteria. Microbiol. Mol. Biol. Rev. 2009, 64, 503–514. [Google Scholar] [CrossRef]
- Claverys, J.P.; Håvarstein, L.S. Cannibalism and fratricide: Mechanisms and raisons d’etre. Nat. Rev. Microbiol. 2007, 5, 219–229. [Google Scholar] [CrossRef]
- Renelli, M.; Matias, V.; Lo, R.Y.; Beveridge, T.J. DNA-containing membrane vesicles of Pseudomonas aeruginosa PAO1 and their genetic transformation potential. Microbiology 2004, 150, 2161–2169. [Google Scholar] [CrossRef]
- Webb, J.S.; Thompson, L.S.; James, S.; Charlton, T.; Tolker-Nielsen, T.; Koch, B.; Givskov, M.K.; Jelleberg, S. Cell death in Pseudomonas aeruginosa biofilm development. J. Bacteriol. 2003, 185, 4585–4592. [Google Scholar] [CrossRef]
- Salgado-Pabón, W.; Du, Y.; Hackett, K.T.; Lyons, K.M.; Arvidson, C.G.; Dillard, J.P. Increased expression of the type IV secretion system in piliated Neisseria gonorrhoeae variants. J. Bacteriol. 2010, 192, 1912–1920. [Google Scholar] [CrossRef]
- Fagerlind, M.G.; Webb, J.S.; Barraud, N.; McDougald, D.; Jansson, A.; Nilsson, P.; Harlén, M.; Kjelleberg, S.; Rice, S.A. Dynamic modelling of cell death during biofilm development. J. Theor. Biol. 2012, 295, 23–36. [Google Scholar] [CrossRef]
- Paczosa, M.K.; Mecsas, J. Klebsiella pneumoniae: Going on the offense with a strong defense. Microbiol. Mol. Biol. Rev. 2016, 80, 629–661. [Google Scholar] [CrossRef]
- Singla, S.; Harjai, K.; Chhibber, S. Artificial Klebsiella pneumoniae biofilm model mimicking in vivo system: altered morphological characteristics and antibiotic resistance. J. Antibiot. 2014, 67, 305–309. [Google Scholar] [CrossRef]
- Stepanović, S.; Ćirković, I.; Ranin, L.; SvabićVlahović, M. Biofilm formation by Salmonella spp. And Listeria monocytogenes on plastic surface. Lett. Appl. Microbiol. 2004, 38, 428–432. [Google Scholar] [CrossRef]
- Shmaefsky, B.R. Artificial urine for laboratory testing. Am. Biol. Teach. 1990, 52, 170–172. [Google Scholar] [CrossRef]
- Wu, J.; Xi, C. Evaluation of different methods for extracting extracellular DNA from biofilm matrix. Appl. Environ. Microbiol. 2009, 75, 5390–5395. [Google Scholar] [CrossRef]
- Madsen, J.S.; Lin, Y.C.; Squyres, G.R.; Price-Whelan, A.; de Santiago Torio, A.; Song, A.; Cornell, W.C.; Sørensen, S.J.; Xavier, J.B.; Dietrich, L.E. Facultative control of matrix production optimizes competitive fitness in Pseudomonas aeruginosa PA14 biofilm models. Appl. Environ. Microbiol. 2015, 81, 8414–8426. [Google Scholar] [CrossRef]
- Accurate Assessment of Microbial Viability by Flow Cytometry 2011, July. BioProbes 65. Available online: https://www.thermofisher.com/in/en/home/references/newsletters-and-journals/bioprobes-journal-of-cell-biology-applications/bioprobes-issues-2011/bioprobes-65-july-2011/live-dead-baclight-meets-the-attune-acoustic-focusing-cytometer.html (accessed on 1 September 2019).






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desai, S.; Sanghrajka, K.; Gajjar, D. High Adhesion and Increased Cell Death Contribute to Strong Biofilm Formation in Klebsiella pneumoniae. Pathogens 2019, 8, 277. https://doi.org/10.3390/pathogens8040277
Desai S, Sanghrajka K, Gajjar D. High Adhesion and Increased Cell Death Contribute to Strong Biofilm Formation in Klebsiella pneumoniae. Pathogens. 2019; 8(4):277. https://doi.org/10.3390/pathogens8040277
Chicago/Turabian StyleDesai, Siddhi, Kinjal Sanghrajka, and Devarshi Gajjar. 2019. "High Adhesion and Increased Cell Death Contribute to Strong Biofilm Formation in Klebsiella pneumoniae" Pathogens 8, no. 4: 277. https://doi.org/10.3390/pathogens8040277
APA StyleDesai, S., Sanghrajka, K., & Gajjar, D. (2019). High Adhesion and Increased Cell Death Contribute to Strong Biofilm Formation in Klebsiella pneumoniae. Pathogens, 8(4), 277. https://doi.org/10.3390/pathogens8040277