Kupffer Cells Survive Plasmodium berghei Sporozoite Exposure and Respond with a Rapid Cytokine Release



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
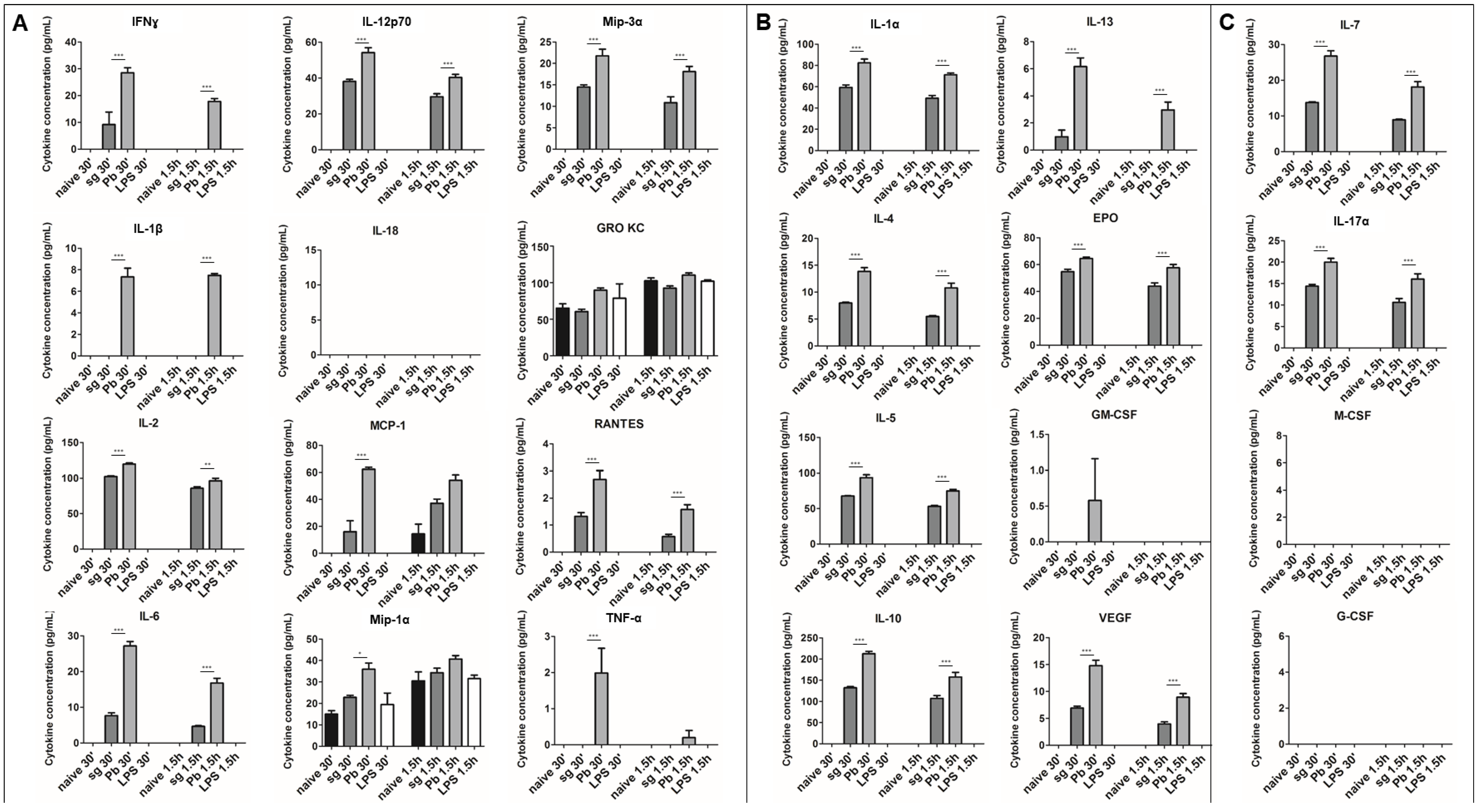
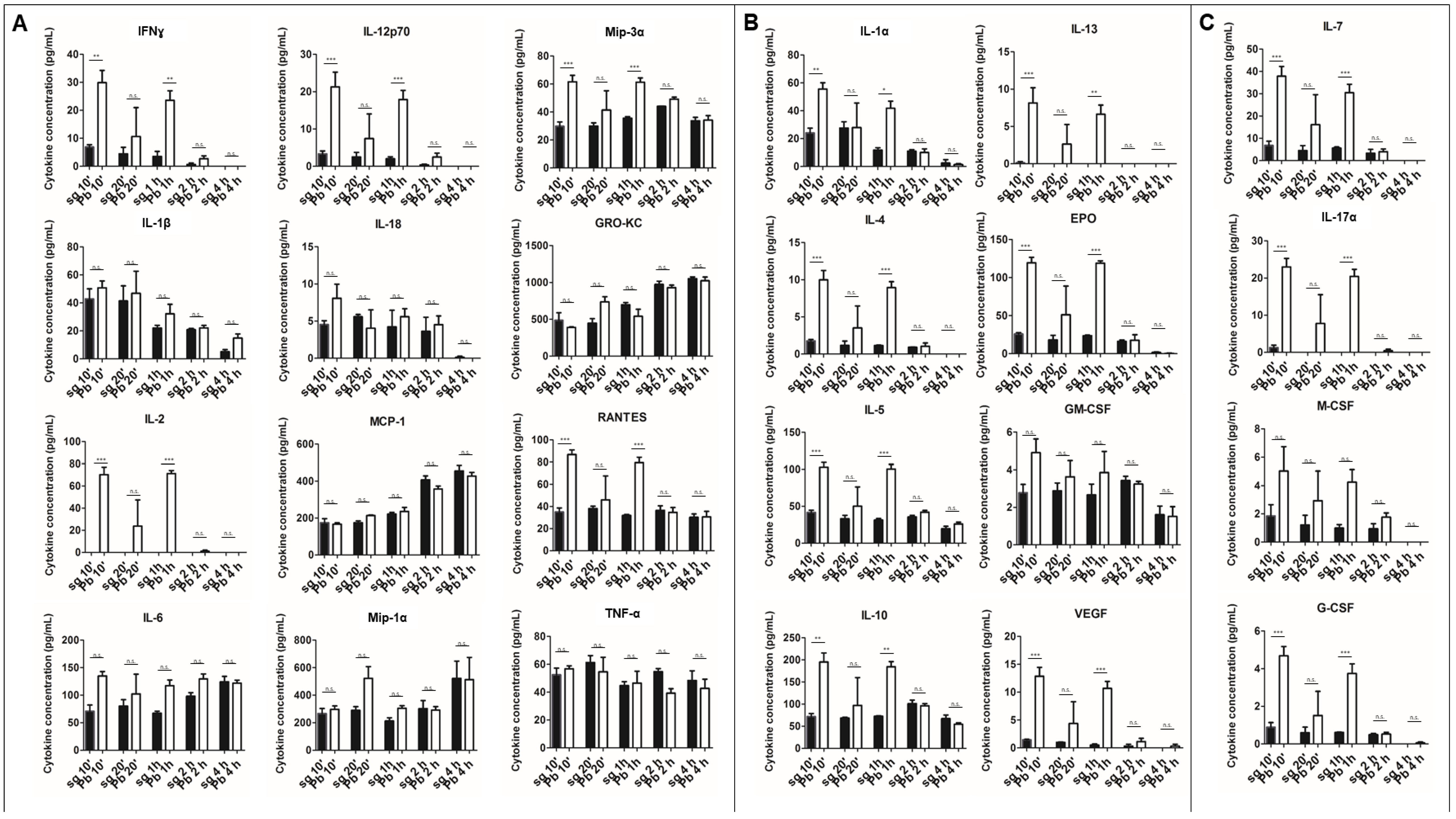
2.1. PRKCs Secrete a Diverse Array of Cytokines in Response to Sporozoite Exposure
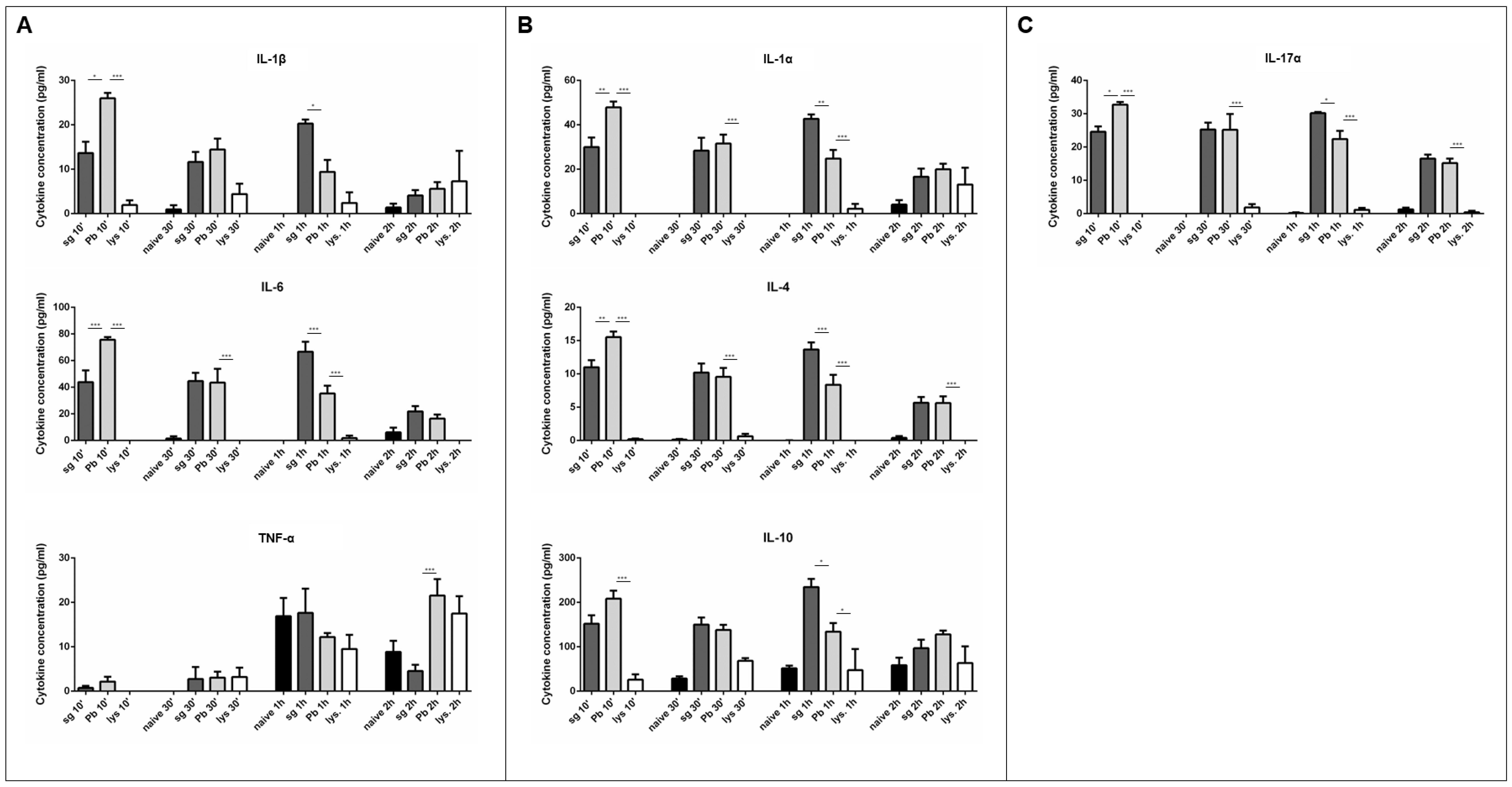
2.2. PRKC Cytokine Secretion is Specific to Live Sporozoite Exposure
2.3. T cells are not Major Contributors to the Observed Cytokine Secretion
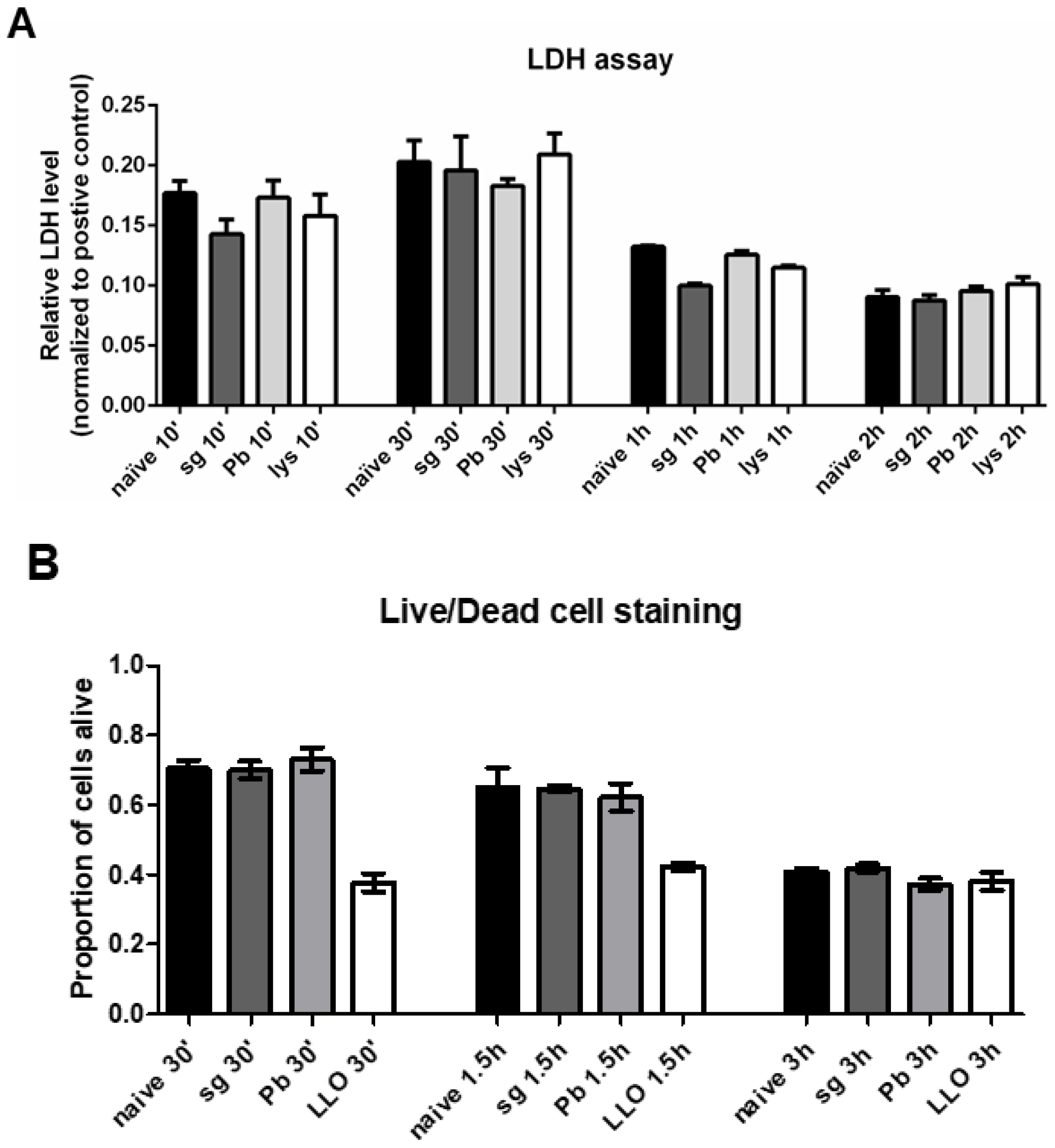
2.4. PRKCs do not Undergo Increased Levels of Cell Death Following Sporozoite Exposure
3. Discussion
4. Materials and Methods
4.1. Cells
4.2. Primary Cell Isolation and Purification
4.3. FACS Analysis for T Cell Quantification
4.4. Sporozoite Generation and Collection and Uninfected Salivary Gland Extract Collection
4.5. Bio-Plex Cytokine Assays
4.6. Lactate Dehydrogenase (LDH) Assay
4.7. Live/Dead Cell Imaging Assay
Supplementary Materials
Author Contributions
Funding
Acknowledgements
Conflicts of Interest
References
- WHO. World Malaria Report 2015; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Yamauchi, L.M.; Coppi, A.; Snounou, G.; Sinnis, P. Plasmodium sporozoites trickle out of the injection site. Cell Microbiol. 2007, 9, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, A.M.; Aly, A.S.; Kappe, S.H. Malaria parasite pre-erythrocytic stage infection: Gliding and hiding. Cell Host Microbe 2008, 4, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Tavares, J.; Formaglio, P.; Thiberge, S.; Mordelet, E.; Van Rooijen, N.; Mevinsky, A.; Menard, R.; Amino, R. Role of host cell traversal by the malaria sporozoite during liver infection. J. Exp. Med. 2013, 210, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.J.; Poucell, S.; Patterson, J. The Liver: An Atlas and Text of Ultrastructural Pathology; Raven Press: New York, USA, 1987; ISBN 9780881673029. [Google Scholar]
- Malakey, D.E.; Johnshon, K.; Ryan, L.; Boorman, G.; Maronpot, R.R. New insights into functional aspects of liver morphology. Toxicol. Pathol. 2005, 33, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.; Zhadkewich, M.; Margolick, J.; Winkelstein, J.; Bulkley, G. Quantitative discrimination of hepatic reticuloendothelial clearance and phagocytic killing. J. Leukoc. Biol. 1994, 55, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Sleyster, E.C.; Knook, D.L. Relation between localization and function of rat liver Kupffer cells. Lab. Investig. 1982, 47, 484–490. [Google Scholar] [PubMed]
- Arai, M.; Peng, X.X.; Currin, R.T.; Thurman, R.G.; Lemasters, J.J. Protection of sinusoidal endothelial cells against storage/reperfusion injury by prostaglandin E2 derived from Kupffer cells. Transplantation 1999, 68, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Crispe, I.N.; Dao, T.; Klugewitz, K.; Mehal, W.Z.; Metz, D.P. The liver as a site of T-cell apoptosis: Graveyard or killing field? Immunol. Rev. 2000, 174, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.X.; Govindarajan, S.; Okamoto, S.; Dennert, G. Fas-mediated apoptosis causes elimination of virus-specific cytotoxic T cells in the virus-infected liver. J. Immunol. 2001, 166, 3035–3041. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K.; Kaisho, T. Toll-like receptors: Critical proteins linking innate and acquired immunity. Nat. Immun. 2001, 2, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Beattie, L.; Peltan, A.; Maroof, A.; Kirby, A.; Brown, N.; Coles, M.; Smith, D.F.; Kaye, P.M. Dynamic imaging of experimental Leishmania donovani-induced hepatic granulomas detects Kupffer cell-restricted antigen presentation to antigen-specific CD8 T cells. PLoS Pathog. 2010, 6, e1000805. [Google Scholar] [CrossRef] [PubMed]
- Seki, S.; Habu, Y.; Kawamura, T.; Takeda, K.; Dobashi, H.; Ohkawa, T.; Hiraide, H. The liver as a crucial organ in the first line of host defense: The roles of Kupffer cells, natural killer (NK) cells and the NK1.1 Ag+ T cells in the T helper 1 immune responses. Immunol. Rev. 2000, 174, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Pradel, G.; Frevert, U. Malaria sporozoites actively enter and pass through rat Kupffer cells prior to hepatocyte invasion. Hepatology 2001, 22, 1154–1165. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, L.A.; Rodo, J.; Rodrigues-Duarte, L.; Vieira de Moraes, L.; Penha-Goncalves, C. HGF secreted by activated Kupffer cells induces apoptosis of Plasmodium-infected hepatocytes. Front. Immunol. 2017, 8, 90. [Google Scholar] [CrossRef] [PubMed]
- Liehl, P.; Meireles, P.; Albuqureque, I.S.; Pinkevych, M.; Baptista, F.; Mota, M.M.; Davenport, M.P.; Prudencio, M. Innate immunity induced by Plasmodium liver infection inhibits malaria reinfections. Infect. Immun. 2015, 83, 1172–1180. [Google Scholar] [CrossRef] [PubMed]
- Liehl, P.; Zuzarte-Luis, V.; Chan, J.; Zillinger, T.; Baptista, F.; Carapau, D.; Konert, M.; Hanson, K.K.; Carret, C.; Lassnig, C.; et al. Host-cell sensors for Plasmodium activate innate immunity against liver-stage infection. Nat. Med. 2014, 20, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Spence, P.J.; Jarra, W.; Levy, P.; Reid, A.J.; Cappell, L.; Brugat, T.; Sanders, M.; Berriman, M.; Langhorne, J. Vector transmission regulates immune control of Plasmodium virulence. Nature 2014, 498, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Klotz, C.; Frevert, U. Plasmodium yoelii sporozoites modulate cytokine profile and induce apoptosis in murine Kupffer cells. Int. J. Parasitol. 2008, 38, 1639–1650. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Gores, G.J. Cellular and molecular mechanisms of liver injury. Gastroenterology 2008, 134, 1641–1654. [Google Scholar] [CrossRef] [PubMed]
- Frevert, U.; Engelmann, S.; Zougbede, S.; Stange, J.; Ng, B.; Matuschewski, K.; Liebes, L.; Yee, H. Intravital observation of Plasmodium berghei sporozoite infection of the liver. PLoS Biol. 2005, 3, e192. [Google Scholar] [CrossRef] [PubMed]
- Mota, M.M.; Hafalla, J.C.; Rodriguez, A. Migration through host cells activates Plasmodium sporozoites for infection. Nat. Med. 2002, 8, 1318–1322. [Google Scholar] [CrossRef] [PubMed]
- Iwashita, M.; Sakoda, H.; Kushiyama, A.; Fujishiro, M.; Ohno, H.; Nakatsu, Y.; Fukushima, T.; Kumamoto, S.; Tsuchiya, Y.; Kikuchi, T.; et al. Valsartan, independently of AT1 receptor or PPARγ, suppresses LPS-induced macrophage activation and improves insulin resistance in cocultured adipocytes. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E286–E296. [Google Scholar] [CrossRef] [PubMed]
- Miletic, A.V.; Graham, D.B.; Montgrain, V.; Fujikawa, K.; Kloeppel, T.; Brim, K.; Weaver, B.; Schreiber, R.; Xavier, R.; Swat, W. Vav proteins control MyD88-dependent oxidative burst. Blood 2007, 109, 3360–3368. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; House, B.L.; Zyzak, M.D.; Richie, T.R.; Gerbasi, V.R. Towards an optimized inhibition of liver stage development assay (ILSDA) for Plasmodium falciparum. Malar. J. 2013, 12, 394. [Google Scholar] [CrossRef] [PubMed]
- Lindner, S.E.; Swearingen, K.E.; Harupa, A.; Vaughan, A.M.; Sinnis, P.; Moritz, R.L.; Kappe, S.H. Total and putative surface proteomics of malaria parasite salivary gland sporozoites. Mol. Cell. Proteom. 2013, 12, 1127–1143. [Google Scholar] [CrossRef] [PubMed]
- Swearingen, K.E.; Lindner, S.E.; Flannery, E.L.; Vaughan, A.M.; Morrison, R.D.; Patrapuvich, R.; Koepfli, C.; Muller, I.; Jex, A.; Moritz, R.L.; et al. Proteogenomic analysis of the total and surface-exposed proteomes of Plasmodium vivax salivary gland sporozoites. PLoS Negl. Trop. Dis. 2017, 11, e0005791. [Google Scholar] [CrossRef] [PubMed]
- Ghayur, T.; Banerjee, S.; Hugunin, M.; Butler, D.; Herzog, L.; Carter, A.; Quintal, L.; Sekut, L.; Talanian, R.; Paskind, M.; et al. Caspase-1 processes IFNγ-inducing factor and regulates LPS-induced IFNγ production. Nature 1997, 386, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Hsu, W.; Wang, C.; Liang, J.; Tsai, M.; Yen, C.; Li, H.; Chiu, S.; Chang, C.; Huang, Y.; et al. A novel strategy for TNFa production by 2-APB induced downregulated SOCE and upregulated HSP70 in O. tsutsugamushi-infected human macrophages. PLoS ONE 2016, 11, e0159299. [Google Scholar]
- Verhoef, P.A.; Kertesy, S.B.; Estacion, M.; Schilling, W.P.; Dubyak, G.R. Maitotoxin induces biphasic interleukin-1b secretion and membrane blebbing in murine macrophages. Mol. Pharmacol. 2004, 66, 909–920. [Google Scholar] [PubMed]
- Karnati, H.K.; Pasupuleti, S.R.; Kandi, R.; Undi, R.B.; Sahu, I.; Kannaki, T.R.; Subbiah, M.; Gutti, R.K. TLR-4 signaling pathway: MyD88 independent pathway up-regulation in chicken breeds upon LPS treatment. Vet. Res. Commun. 2015, 39, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Lefebvre, A.T.; Horuzsko, A. Kupffer cell metabolism and function. J. Enzymol. Metab. 2015, 1, 101. [Google Scholar] [PubMed]
- Seki, E.; Tsutsui, H.; Nakano, H.; Tsuji, N.; Hoshino, K.; Adachi, O.; Adachi, K.; Futatsugi, S.; Kuida, K.; Taeuchi, O.; et al. Lipopolysaccharide-induced IL-18 secretion from murine Kupffer cells independently of myeloid differentiation factor 88 that is critically involved in induction of production of IL-12 and IL-1beta. J. Immunol. 2001, 166, 2651–2657. [Google Scholar] [CrossRef] [PubMed]
- Lord, K.A.; Hoffman-Liebermann, B.; Liebermann, D.A. Nucleotide sequence and expression of a cDNA encoding MyD88, a novel myeloid differentiation primary response gene induced by IL6. Oncogene 1990, 5, 1095–1097. [Google Scholar] [PubMed]
- Naik, R.S.; Branch, O.H.; Woods, A.S.; Vjaykumar, M.; Perkins, D.J.; Nahlen, B.L.; Lal, A.A.; Cotter, R.J.; Costello, C.E.; Ockenhouse, C.F.; et al. Glycosylphosphatidylinositol anchors of Plasmodium falciparum: Molecular characterization and naturally elicited antibody response that may provide immunity to malaria pathogenesis. J. Exp. Med. 2000, 192, 1563–1576. [Google Scholar] [CrossRef] [PubMed]
- Bautista, A.P. Chronic alcohol intoxication primes Kupffer cells and endothelial cells for enhanced CC-chemokine production and concomitantly suppresses phagocytosis and chemotaxis. Front. Biosci. 2002, 7, 117–125. [Google Scholar] [CrossRef]
- Ramadori, P.; Ahmad, G.; Ramadori, G. Cellular and molecular mechanisms regulating the hepatic erythropoietin expression during acute-phase response: A role for IL-6. Lab. Investig. 2010, 90, 1306–1324. [Google Scholar] [CrossRef] [PubMed]
- Tanikawa, K.; Ueno, T. (Eds.) Liver Diseases and Hepatic Sinusoidal Cells; Springer Publishing Co.: Tokyo, Japan, 1999; ISBN 978-4-431-67935-6. [Google Scholar]
- Oster, W.; Lindemann, A.; Mertelsmann, R.; Herrmann, F. Production of macrophage-, granulocyte-, granulocyte-macrophage- and multi-colony stimulating factor by peripheral blood cells. Eur. J. Immunol. 1989, 19, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Grinchuk, V.; Urban, J.F., Jr.; Bohl, J.; Sun, R.; Notari, L.; Yan, S.; Ramalingam, T.; Keegan, A.D.; Wynn, T.A.; et al. Macrophages as IL-25/IL-33-responsive cells play an important role in the induction of Type 2 immunity. PLoS ONE 2013, 8, e59441. [Google Scholar] [CrossRef] [PubMed]
- Sawa, Y.; Arima, Y.; Ogura, H.; Kitabayashi, C.; Jiang, J.J.; Fukushima, T.; Kamimura, D.; Hirano, T.; Murakami, M. Hepatic interleukin-7 expression regulated T cell responses. Immunity 2009, 30, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Sierro, F.; Evrard, M.; Rizzetto, S.; Melino, M.; Mitchell, A.J.; Florido, M.; Beattie, L.; Walters, S.B.; Tay, S.S.; Lu, B.; et al. A liver capsular network of monocyte-derived macrophages restricts hepatic dissemination of intraperitoneal bacteria by neutrophil recruitment. Immunity 2017, 47, 374–388. [Google Scholar] [CrossRef] [PubMed]
- Usynin, I.; Klotz, C.; Frevert, U. Malaria circumsporozoite protein inhibits the respiratory burst in Kupffer cells. Cell Microbiol. 2007, 9, 2610–2628. [Google Scholar] [CrossRef] [PubMed]
- Ishino, T.; Chinzel, Y.; Yuda, M. A Plasmodium sporozoite protein with a membrane attack complex domain is required for breaching the liver sinusoidal cell layer prior to hepatocyte infection. Cell Microbiol. 2005, 7, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Walbrun, P.; Hellerbrand, C.; Weiss, T.S.; Netter, S.; Neumaier, D.; Gaebele, E.; Wiest, R.; Schoelmerich, J.; Froh, M. Characterization of rat and human Kupffer cells after cryopreservation. Cryobiology 2007, 54, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Butler, N.S.; Schmidt, N.W.; Harty, J.T. Differential effector pathways regulate memory CD8 T cell immunity against Plasmodium berghei versus P. yoelii sporozoites. J. Immunol. 2010, 184, 2528–2538. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Wada, T.; Hoshino, S.; Uchikura, K.; Klein, A.S. Immunomodulatory role of Kupffer cell in liver allografts. Comp. Hepatol. 2004, 3, S32. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sinnis, P.; De La Vega, P.; Coppi, A.; Krzych, U.; Mota, M.M. Quantification of sporozoite invasion, migration, and development by microscopy and flow cytometry. Methods Mol. Biol. 2013, 923, 385–400. [Google Scholar] [PubMed]
- Cummings, B.S.; Wills, L.P.; Schnellmann, R.G. Measurement of cell death in mammalian cells. Curr. Protoc. Pharmacol. 2004, 12. [Google Scholar] [CrossRef]
- Kroemer, G.; Martin, S.J. Caspase-independent cell death. Nat. Med. 2005, 11, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Leist, M.; Jaattela, M. Four deaths and a funeral: From caspases to alternative mechanisms. Nat. Rev. 2001, 2, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Wlodkowic, D.; Telford, W.; Skommer, J.; Darzynkiewicz, Z. Apoptosis and beyond: Cytometry in studies of programmed cell death. Methods Cell Biol. 2011, 103, 55–98. [Google Scholar] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tweedell, R.E.; Qi, L.; Sun, Z.; Dinglasan, R.R. Kupffer Cells Survive Plasmodium berghei Sporozoite Exposure and Respond with a Rapid Cytokine Release. Pathogens 2018, 7, 91. https://doi.org/10.3390/pathogens7040091
Tweedell RE, Qi L, Sun Z, Dinglasan RR. Kupffer Cells Survive Plasmodium berghei Sporozoite Exposure and Respond with a Rapid Cytokine Release. Pathogens. 2018; 7(4):91. https://doi.org/10.3390/pathogens7040091
Chicago/Turabian StyleTweedell, Rebecca E., Le Qi, Zhaoli Sun, and Rhoel R. Dinglasan. 2018. "Kupffer Cells Survive Plasmodium berghei Sporozoite Exposure and Respond with a Rapid Cytokine Release" Pathogens 7, no. 4: 91. https://doi.org/10.3390/pathogens7040091
APA StyleTweedell, R. E., Qi, L., Sun, Z., & Dinglasan, R. R. (2018). Kupffer Cells Survive Plasmodium berghei Sporozoite Exposure and Respond with a Rapid Cytokine Release. Pathogens, 7(4), 91. https://doi.org/10.3390/pathogens7040091