Modelling a Silent Epidemic: A Review of the In Vitro Models of Latent Tuberculosis



{kind=link}
{kind=link}
Abstract
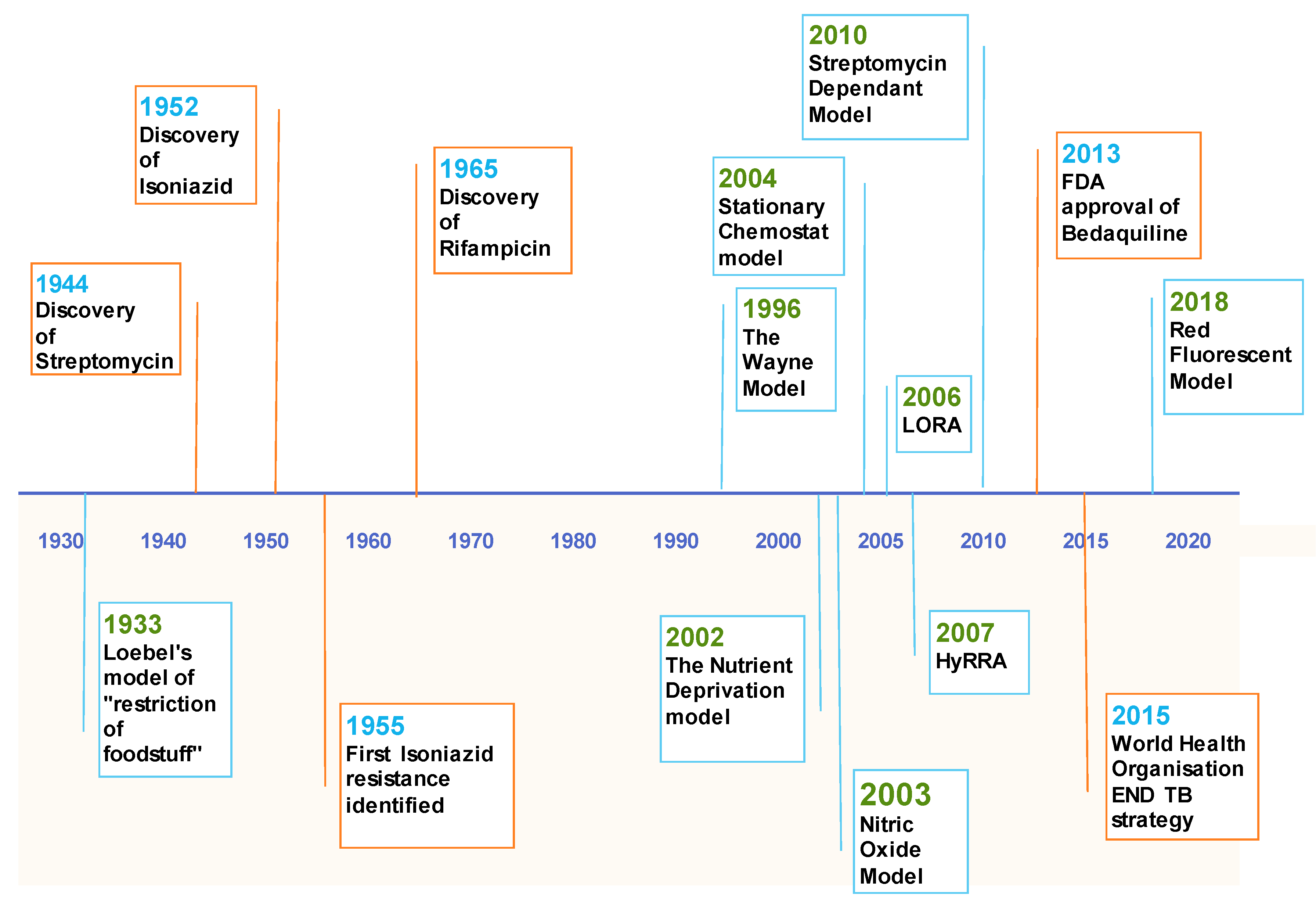
1. Introduction
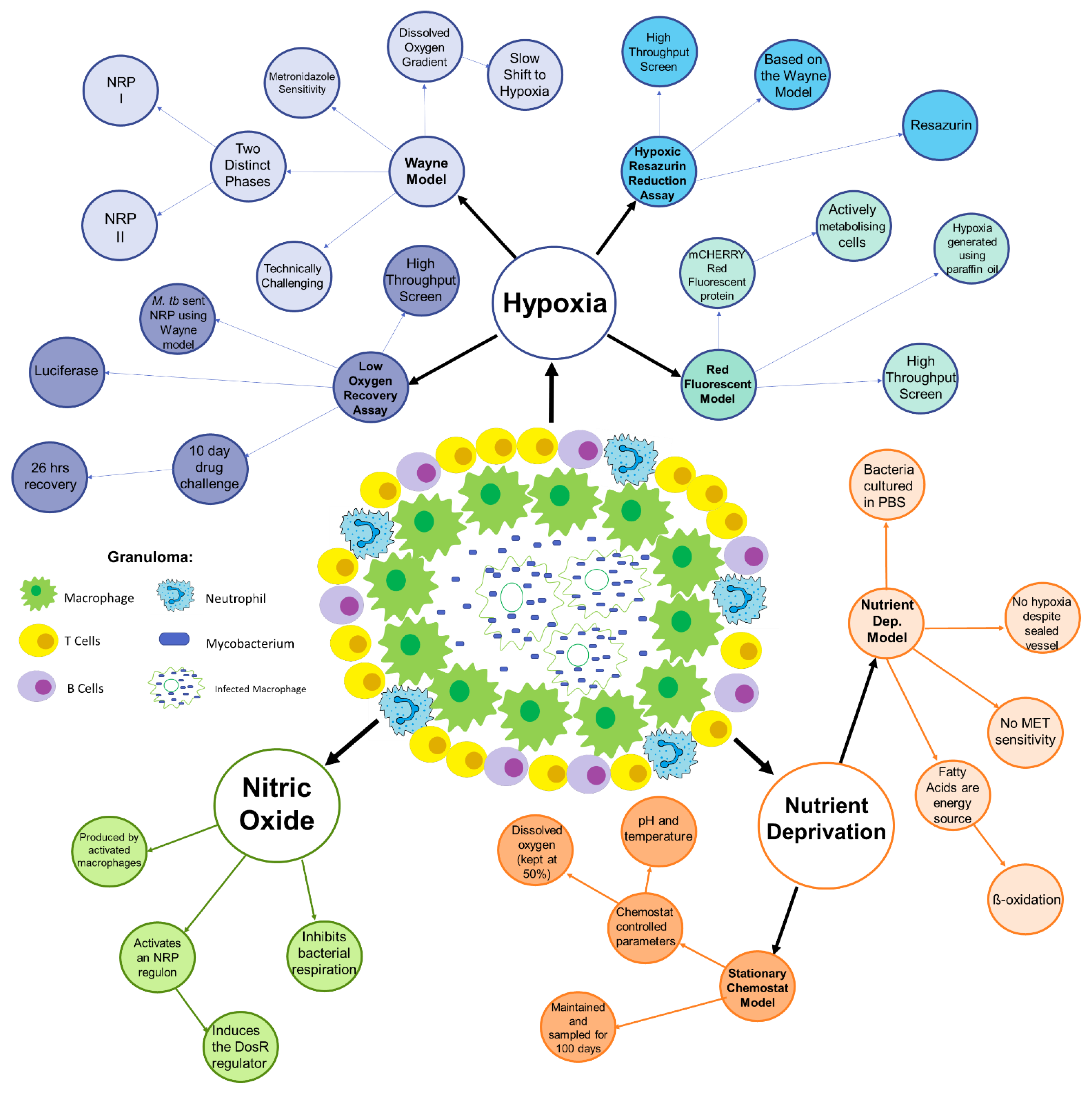
2. Conditions within the Granuloma
3. Hypoxia
3.1. The Wayne Model
3.2. Hypoxic Resazurin Reduction Assay (HyRRA)
3.3. Low Oxygen Recovery Assay (LORA)
3.4. Red Fluorescent Protein (RFP) Model
4. Nutrient Deprivation and Selective Carbon Sources
4.1. The Nutrient Deprivation Model
4.2. Stationary Chemostat Model
5. Nitric Oxide
6. Streptomycin Dependent
7. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Global Tuberculosis Report 2017; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Smith, I. Mycobacterium tuberculosis pathogenesis and molecular determinants of virulence. Clin. Microbiol. Rev. 2003, 16, 463–496. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, V.; Wiegeshaus, E.H.; Taylor, B.T.; Smith, D.W. Pathogenesis of tuberculosis: Pathway to apical localization. Tuber. Lung Dis. 1994, 75, 168–178. [Google Scholar] [CrossRef]
- Kang, D.D.; Lin, Y.; Moreno, J.-R.; Randall, T.D.; Khader, S.A. Profiling early lung immune responses in the mouse model of tuberculosis. PLoS ONE 2011, 6, e16161. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.J.; Linas, B.; Trevejo-Nuñez, G.J.; Kincaid, E.; Tamura, T.; Takatsu, K.; Ernst, J.D. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J. Immunol. 2007, 179, 2509–2519. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, A. Some clinical features of tuberculosis. 1. Incubation period. Acta Tuberc. Scand. 1950, 24, 311. [Google Scholar] [PubMed]
- Russell, D.G. Who puts the tubercle in tuberculosis? Nat. Rev. Microbiol. 2007, 5, 39. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, L. Revisiting the role of the granuloma in tuberculosis. Nat. Rev. Immunol. 2012, 12, 352. [Google Scholar] [CrossRef] [PubMed]
- Guirado, E.; Schlesinger, L. Modeling the Mycobacterium tuberculosis granuloma–the critical battlefield in host immunity and disease. Front. Immunol. 2013, 4, 98. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.D. The immunological life cycle of tuberculosis. Nat. Rev. Immunol. 2012, 12, 581. [Google Scholar] [CrossRef] [PubMed]
- Knechel, N.A. Tuberculosis: Pathophysiology, clinical features, and diagnosis. Crit. Care Nurse 2009, 29, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Lönnroth, K.; Castro, K.G.; Chakaya, J.M.; Chauhan, L.S.; Floyd, K.; Glaziou, P.; Raviglione, M.C. Tuberculosis control and elimination 2010–50: Cure, care, and social development. Lancet 2010, 375, 1814–1829. [Google Scholar] [CrossRef]
- Espinal, M.A. The global situation of MDR-TB. Tuberculosis 2003, 83, 44–51. [Google Scholar] [CrossRef]
- WHO. The End TB Strategy; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations, 2016; HM Government and Welcome Trust: London, UK, 2018. [Google Scholar]
- Barry, C.E., 3rd; Boshoff, H.I.; Dartois, V.; Dick, T.; Ehrt, S.; Flynn, J.; Schnappinger, D.; Wilkinson, R.J.; Young, D. The spectrum of latent tuberculosis: Rethinking the biology and intervention strategies. Nat. Rev. Microbiol. 2009, 7, 845. [Google Scholar] [CrossRef] [PubMed]
- Wayne, L.G.; Sohaskey, C.D. Nonreplicating persistence of Mycobacterium tuberculosis. Ann. Rev. Microbiol. 2001, 55, 139–163. [Google Scholar] [CrossRef] [PubMed]
- Esmail, H.; Barry, C.E.; Young, D.B.; Wilkinson, R.J. The ongoing challenge of latent tuberculosis. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130437. [Google Scholar] [CrossRef] [PubMed]
- Getahun, H.; Matteelli, A.; Chaisson, R.E.; Raviglione, M. Latent Mycobacterium tuberculosis Infection. N. Engl. J. Med. 2015, 372, 2127–2135. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.E.; Pandey, A.K.; Gilmore, S.A.; Mizrahi, V.; McKinney, J.D.; Bertozzi, C.R.; Sassetti, C.M. Cholesterol catabolism by mycobacterium tuberculosis requires transcriptional and metabolic adaptations. Chem. Biol. 2012, 19, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Soto-Ramirez, M.D.; Aguilar-Ayala, D.A.; Garcia-Morales, L.; Rodriguez-Peredo, S.M.; Badillo-Lopez, C.; Rios-Muñiz, D.E.; Meza-Segura, M.A.; Rivera-Morales, G.Y.; Leon-Solis, L.; Cerna-Cortes, J.F.; et al. Cholesterol plays a larger role during Mycobacterium tuberculosis in vitro dormancy and reactivation than previously suspected. Tuberculosis 2017, 103, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Elías, E.J.; Upton, A.M.; Cherian, J.; McKinney, J.D. Role of the methylcitrate cycle in Mycobacterium tuberculosis metabolism, intracellular growth, and virulence. Mol. Microbiol. 2006, 60, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Wayne, L.G. Dynamics of submerged growth of Mycobacterium tuberculosis under aerobic and microaerophilic conditions. Am. Rev. Respir. Dis. 1976, 114, 807–811. [Google Scholar] [PubMed]
- Huebner, R.E.; Schein, M.F.; Bass, J.B., Jr. The tuberculin skin test. Clin. Infect. Dis. 1993, 17, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Lordi, G.M.; Reichman, L.B. Tuberculin skin testing. In Tuberculosis; Springer: Berlin, Germany, 1988; pp. 33–38. [Google Scholar]
- Houben, R.; Dodd, P.J. The global burden of latent tuberculosis infection: A re-estimation using mathematical modelling. PLoS Med. 2016, 13, e1002152. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, A.M.; Spigelman, M. Challenges in tuberculosis drug research and development. Nat. Med. 2007, 13, 290. [Google Scholar] [CrossRef] [PubMed]
- Puissegur, M.-P.; Botanch, C.; Duteyrat, J.-L.; Delsol, G.; Caratero, C.; Altare, F. An in vitro dual model of mycobacterial granulomas to investigate the molecular interactions between mycobacteria and human host cells. Cell. Microbiol. 2004, 6, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Voskuil, M.I.; Visconti, K.C.; Schoolnik, G.K. Mycobacterium tuberculosis gene expression during adaptation to stationary phase and low-oxygen dormancy. Tuberculosis 2004, 84, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Brzostek, A.; Pawelczyk, J.; Rumijowska-Galewicz, A.; Dziadek, B.; Dziadek, J. Mycobacterium tuberculosis is able to accumulate and utilize cholesterol. J. Bacteriol. 2009, 191, 6584–6591. [Google Scholar] [CrossRef] [PubMed]
- Betts, J.; Lukey, P.; Robb, L.; McAdam, R.; Duncan, K. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 2002, 43, 717–731. [Google Scholar] [CrossRef] [PubMed]
- Loebel, R.O.; Shorr, E.; Richardson, H.B. The influence of adverse conditions upon the respiratory metabolism and growth of human tubercle bacilli. J. Bacteriol. 1933, 26, 167–200. [Google Scholar] [PubMed]
- Nyka, W. Studies on the effect of starvation on mycobacteria. Infect. Immun. 1974, 9, 843–850. [Google Scholar] [PubMed]
- Nathan, C.; Ehrt, S. Nitric oxide in tuberculosis. Tuberculosis 2004, 84, 215–235. [Google Scholar]
- Wayne, L.G.; Hayes, L.G. An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect. Immun. 1996, 64, 2062–2069. [Google Scholar] [PubMed]
- Boon, C.; Li, R.; Qi, R.; Dick, T. Proteins of Mycobacterium bovis BCG induced in the Wayne dormancy model. J. Bacteriol. 2001, 183, 2672–2676. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Jhamb, S.S.; Singh, P.P. Models of latent tuberculosis: Their salient features, limitations, and development. J. Lab. Phys. 2011, 3, 75–79. [Google Scholar] [CrossRef]
- Wayne, L.G.; Sramek, H.A. Metronidazole is bactericidal to dormant cells of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1994, 38, 2054–2058. [Google Scholar] [CrossRef] [PubMed]
- Wayne, L.G. In vitro model of hypoxically induced nonreplicating persistence of Mycobacterium tuberculosis. In Mycobacterium Tuberculosis Protocols; Springer: Berlin, Germany, 2001; pp. 247–269. [Google Scholar]
- Klinkenberg, L.G.; Sutherland, L.A.; Bishai, W.R.; Karakousis, P.C. Metronidazole lacks activity against Mycobacterium tuberculosis in an in vivo hypoxic granuloma model of latency. J. Infect. Dis. 2008, 198, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Hoff, D.R.; Caraway, M.L.; Brooks, E.J.; Driver, E.R.; Ryan, G.J.; Peloquin, C.A.; Orme, I.M.; Basaraba, R.J.; Lenaerts, A.J. Metronidazole lacks antibacterial activity in guinea pigs infected with mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2008, 52, 4137–4140. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Alnimr, A.M. Dormancy models for Mycobacterium tuberculosis: A minireview. Braz. J. Microbiol. 2015, 46, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Broach, J.R.; Thorner, J. High-throughput screening for drug discovery. Nature 1996, 384, 14–16. [Google Scholar] [PubMed]
- Palomino, J.-C.; Martin, A.; Camacho, M.; Guerra, H.; Swings, J.; Portaels, F. Resazurin microtiter assay plate: Simple and inexpensive method for detection of drug resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2002, 46, 2720–2722. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.; Franzblau, S.G. Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob. Agents Chemother. 1997, 41, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Sarker, S.D.; Nahar, L.; Kumarasamy, Y. Microtitre plate-based antibacterial assay incorporating resazurin as an indicator of cell growth, and its application in the in vitro antibacterial screening of phytochemicals. Methods 2007, 42, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Taneja, N.K.; Tyagi, J.S. Resazurin reduction assays for screening of anti-tubercular compounds against dormant and actively growing Mycobacterium tuberculosis, Mycobacterium bovis BCG and Mycobacterium smegmatis. J. Antimicrob. Chemother. 2007, 60, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Murugasu-Oei, B.; Dick, T. Bactericidal activity of nitrofurans against growing and dormant Mycobacterium bovis BCG. J. Antimicrob. Chemother. 2000, 46, 917–919. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Warit, S.; Wan, B.; Hwang, C.H.; Pauli, G.F.; Franzblau, S.G. Low-oxygen-recovery assay for high-throughput screening of compounds against nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2007, 51, 1380–1385. [Google Scholar] [CrossRef] [PubMed]
- Snewin, V.A.; Gares, M.-P.; ÓGaora, P.; Hasan, Z.; Brown, I.N.; Young, D.B. Assessment of immunity to mycobacterial infection with luciferase reporter constructs. Infect. Immun. 1999, 67, 4586–4593. [Google Scholar] [PubMed]
- Duncan, S.; Glover, L.A.; Killham, K.; Prosser, J.I. Luminescence-based detection of activity of starved and viable but nonculturable bacteria. Appl. Environ. Microbiol. 1994, 60, 1308–1316. [Google Scholar] [PubMed]
- Zhang, J.-H.; Chung, T.D.; Oldenburg, K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Timmins, G.S.; Deretic, V. Mechanisms of action of isoniazid. Mol. Microbiol. 2006, 62, 1220–1227. [Google Scholar] [CrossRef] [PubMed]
- Heifets, L.; Simon, J.; Pham, V. Capreomycin is active against non-replicating M. tuberculosis. Ann. Clin. Microbiol. Antimicrob. 2005, 4, 6. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gumbo, T.; Louie, A.; Deziel, M.R.; Parsons, L.M.; Salfinger, M.; Drusano, G.L. Selection of a moxifloxacin dose that suppresses drug resistance in Mycobacterium tuberculosis, by use of an in vitro pharmacodynamic infection model and mathematical modeling. J. Infect. Dis. 2004, 190, 1642–1651. [Google Scholar] [CrossRef] [PubMed]
- Bonnett, S.A.; Dennison, D.; Files, M.; Bajpai, A.; Parish, T. A class of hydrazones are active against non-replicating Mycobacterium tuberculosis. PLoS ONE 2018, 13, e0198059. [Google Scholar] [CrossRef] [PubMed]
- Bonnett, S.A.; Ollinger, J.; Chandrasekera, S.; Florio, S.; O’Malley, T.; Files, M.; Jee, J.-A.; Ahn, J.; Casey, A.; Ovechkina, Y. A target-based whole cell screen approach to identify potential inhibitors of Mycobacterium tuberculosis signal peptidase. ACS Infect. Dis. 2016, 2, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-j.; Petrofsky, M.; Bermudez, L.E. Mycobacterium tuberculosis uptake by recipient host macrophages is influenced by environmental conditions in the granuloma of the infectious individual and is associated with impaired production of interleukin-12 and tumor necrosis factor Alpha. Infect. Immun. 2002, 70, 6223–6230. [Google Scholar] [CrossRef] [PubMed]
- Changsen, C.; Franzblau, S.G.; Palittapongarnpim, P. Improved green fluorescent protein reporter gene-based microplate screening for antituberculosis compounds by utilizing an acetamidase promoter. Antimicrob. Agents Chemother. 2003, 47, 3682–3687. [Google Scholar] [CrossRef] [PubMed]
- Yeware, A.; Sarkar, D. Novel red fluorescence protein based microplate assay for drug screening against dormant Mycobacterium tuberculosis by using paraffin. Tuberculosis 2018, 110, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Carroll, P.; Schreuder, L.J.; Muwanguzi-Karugaba, J.; Wiles, S.; Robertson, B.D.; Ripoll, J.; Ward, T.H.; Bancroft, G.J.; Schaible, U.E.; Parish, T. Sensitive detection of gene expression in mycobacteria under replicating and non-replicating conditions using optimized far-red reporters. PLoS ONE 2010, 5, e9823. [Google Scholar] [CrossRef] [PubMed]
- Parish, T.; Stoker, N.G. Electroporation of mycobacteria. In Mycobacteria Protocols; Springer: Berlin, Germany, 1998; pp. 129–144. [Google Scholar]
- Hugh, R.; Leifson, E. The taxonomic significance of fermentative versus oxidative metabolism of carbohydrates by various Gram negative bacteria. J. Bacteriol. 1953, 66, 24–26. [Google Scholar] [PubMed]
- Gomez, J.E.; McKinney, J.D.M. M. tuberculosis persistence, latency, and drug tolerance. Tuberculosis 2004, 84, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Lee, H.S.; Franzblau, S. Microplate alamar blue assay (MABA) and low oxygen recovery assay (LORA) for Mycobacterium tuberculosis. In Mycobacteria Protocols; Springer: Berlin, Germany, 2015; pp. 281–292. [Google Scholar]
- Murphy, D.J.; Brown, J.R. Identification of gene targets against dormant phase Mycobacterium tuberculosis infections. BMC Infect. Dis. 2007, 7, 84. [Google Scholar] [CrossRef] [PubMed]
- Loebel, R.O.; Shorr, E.; Richardson, H.B. The influence of foodstuffs upon the respiratory metabolism and growth of human tubercle bacilli. J. Bacteriol. 1933, 26, 139–166. [Google Scholar] [PubMed]
- Sarathy, J.; Dartois, V.; Dick, T.; Gengenbacher, M. Reduced drug uptake in phenotypically resistant nutrient-starved non-replicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2013. [Google Scholar] [CrossRef] [PubMed]
- Manabe, Y.C.; Bishai, W.R. Latent Mycobacterium tuberculosis–persistence, patience, and winning by waiting. Nat. Med. 2000, 6, 1327. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Coates, A.R.M. Transcription of two sigma 70 homologue genes, sigA and sigB, in stationary-phase Mycobacterium tuberculosis. J. Bacteriol. 1999, 181, 469–476. [Google Scholar] [PubMed]
- Manganelli, R.; Dubnau, E.; Tyagi, S.; Kramer, F.R.; Smith, I. Differential expression of 10 sigma factor genes in Mycobacterium tuberculosis. Mol. Microbiol. 1999, 31, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Hampshire, T.; Soneji, S.; Bacon, J.; James, B.W.; Hinds, J.; Laing, K.; Stabler, R.A.; Marsh, P.D.; Butcher, P.D. Stationary phase gene expression of Mycobacterium tuberculosis following a progressive nutrient depletion: A model for persistent organisms? Tuberculosis 2004, 84, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Neijssel, O.; Tempest, D. Bioenergetic aspects of aerobic growth of Klebsiella aerogenes NCTC 418 in carbon-limited and carbon-sufficient chemostat culture. Arch. Microbiol. 1976, 107, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Tuomanen, E.; Cozens, R.; Tosch, W.; Zak, O.; Tomasz, A. The rate of killing of Escherichia coli byβ-lactam antibiotics is strictly proportional to the rate of bacterial growth. Microbiology 1986, 132, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Van Andel, J.; Zoutberg, G.; Crabbendam, P.; Breure, A. Glucose fermentation by Clostridium butyricum grown under a self generated gas atmosphere in chemostat culture. Appl. Microbiol. Biotechnol. 1985, 23, 21–26. [Google Scholar] [CrossRef]
- Vulić, M.; Kolter, R. Evolutionary Cheating in Escherichia coli stationary phase cultures. Genetics 2001, 158, 519–526. [Google Scholar] [PubMed]
- Wiker, H.G.; Harboe, M.; Nagai, S. A localization index for distinction between extracellular and intracellular antigens of Mycobacterium tuberculosis. Microbiology 1991, 137, 875–884. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hernandez-Pando, R.; Jeyanathan, M.; Mengistu, G.; Aguilar, D.; Orozco, H.; Harboe, M.; Rook, G.; Bjune, G. Persistence of DNA from Mycobacterium tuberculosis in superficially normal lung tissue during latent infection. Lancet 2000, 356, 2133–2138. [Google Scholar] [CrossRef]
- Corper, H.; Cohn, M.L. The viability and virulence of old cultures of tubercle bacilli. Studies on twelve-year broth cultures maintained at incubator temperature. Am. Rev. Tuberc. Pulm. Dis. 1933, 28, 856–874. [Google Scholar]
- Lyon, R.H.; Lichstein, H.C.; Hall, W.H. Effect of Tween 80 on the growth of tubercle bacilli in aerated cultures. J. Bacteriol. 1963, 86, 280–284. [Google Scholar] [PubMed]
- Mizuno, S.; Tsukamura, M. Utilization of tween 80 as carbon source for growth of slowly growing mycobacteria. Kekkaku (Tuberculosis) 1978, 53, 537–540. [Google Scholar] [CrossRef]
- Brown, G.C. Regulation of mitochondrial respiration by nitric oxide inhibition of cytochrome c oxidase. Biochim. Biophys. Acta (BBA) Bioenerg. 2001, 1504, 46–57. [Google Scholar] [CrossRef]
- Flynn, J.L.; Chan, J. Tuberculosis: Latency and reactivation. Infect. Immun. 2001, 69, 4195–4201. [Google Scholar] [CrossRef] [PubMed]
- Voskuil, M.I.; Schnappinger, D.; Visconti, K.C.; Harrell, M.I.; Dolganov, G.M.; Sherman, D.R.; Schoolnik, G.K. Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J. Exp. Med. 2003, 198, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Boon, C.; Dick, T. Mycobacterium bovis BCG response regulator essential for hypoxic dormancy. J. Bacteriol. 2002, 184, 6760–6767. [Google Scholar] [CrossRef] [PubMed]
- Sherman, D.R.; Voskuil, M.; Schnappinger, D.; Liao, R.; Harrel, M.I.; Schoolnik, G.K. Regulation of the Mycobacterium tuberculosis hypoxic response gene encoding alpha-chrystallin. Proc. Natl. Acad. Sci. USA 2001, 98, 7534–7539. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Crane, D.D.; Simpson, R.M.; Zhu, Y.; Hickey, M.J.; Sherman, D.R.; Barry, C.E. The 16-kDa α-crystallin (Acr) protein of Mycobacterium tuberculosis is required for growth in macrophages. Proc. Natl. Acad. Sci. USA 1998, 95, 9578–9583. [Google Scholar] [CrossRef] [PubMed]
- Wayne, L.; Hayes, L. Nitrate reduction as a marker for hypoxic shiftdown of Mycobacterium tuberculosis. Tuber. Lung Dis. 1998, 79, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Garbe, T.; Hibler, N.; Deretic, V. Response to reactive nitrogen intermediates in Mycobacterium tuberculosis: Induction of the 16-Kilodalton α-Crystallin Homolog by exposure to nitric oxide donors. Infect. Immun. 1999, 67, 460–465. [Google Scholar] [PubMed]
- Hashimoto, T. Experimental studies on the mechanism of infection and immunity in tuberculosis from the analytical standpoint of streptomycin-dependent tubercle bacilli. 1. Isolation and biological characteristics of a streptomycin-dependent mutant, and effect of streptomycin administration on its pathogenicity in guinea-pigs. Kekkaku [Tuberculosis] 1955, 30, 4. [Google Scholar] [PubMed]
- Sala, C.; Dhar, N.; Hartkoorn, R.C.; Zhang, M.; Ha, Y.H.; Schneider, P.; Cole, S.T. Simple model for testing drugs against nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2010, 54, 4150–4158. [Google Scholar] [CrossRef] [PubMed]
- Dawson, R.; Diacon, A.H.; Everitt, D.; van Niekerk, C.; Donald, P.R.; Burger, D.A.; Schall, R.; Spigelman, M.; Conradie, A.; Eisenach, K. Efficiency and safety of the combination of moxifloxacin, pretomanid (PA-824), and pyrazinamide during the first 8 weeks of antituberculosis treatment: A phase 2b, open-label, partly randomised trial in patients with drug-susceptible or drug-resistant pulmonary tuberculosis. Lancet 2015, 385, 1738–1747. [Google Scholar] [PubMed]
- Murray, S.; Mendel, C.; Spigelman, M. TB Alliance regimen development for multidrug-resistant tuberculosis. Int. J. Tuberc. Lung Dis. 2016, 20, S38–S41. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gibson, S.E.R.; Harrison, J.; Cox, J.A.G. Modelling a Silent Epidemic: A Review of the In Vitro Models of Latent Tuberculosis. Pathogens 2018, 7, 88. https://doi.org/10.3390/pathogens7040088
Gibson SER, Harrison J, Cox JAG. Modelling a Silent Epidemic: A Review of the In Vitro Models of Latent Tuberculosis. Pathogens. 2018; 7(4):88. https://doi.org/10.3390/pathogens7040088
Chicago/Turabian StyleGibson, Savannah E.R., James Harrison, and Jonathan A.G. Cox. 2018. "Modelling a Silent Epidemic: A Review of the In Vitro Models of Latent Tuberculosis" Pathogens 7, no. 4: 88. https://doi.org/10.3390/pathogens7040088
APA StyleGibson, S. E. R., Harrison, J., & Cox, J. A. G. (2018). Modelling a Silent Epidemic: A Review of the In Vitro Models of Latent Tuberculosis. Pathogens, 7(4), 88. https://doi.org/10.3390/pathogens7040088