SseL Deubiquitinates RPS3 to Inhibit Its Nuclear Translocation


Abstract
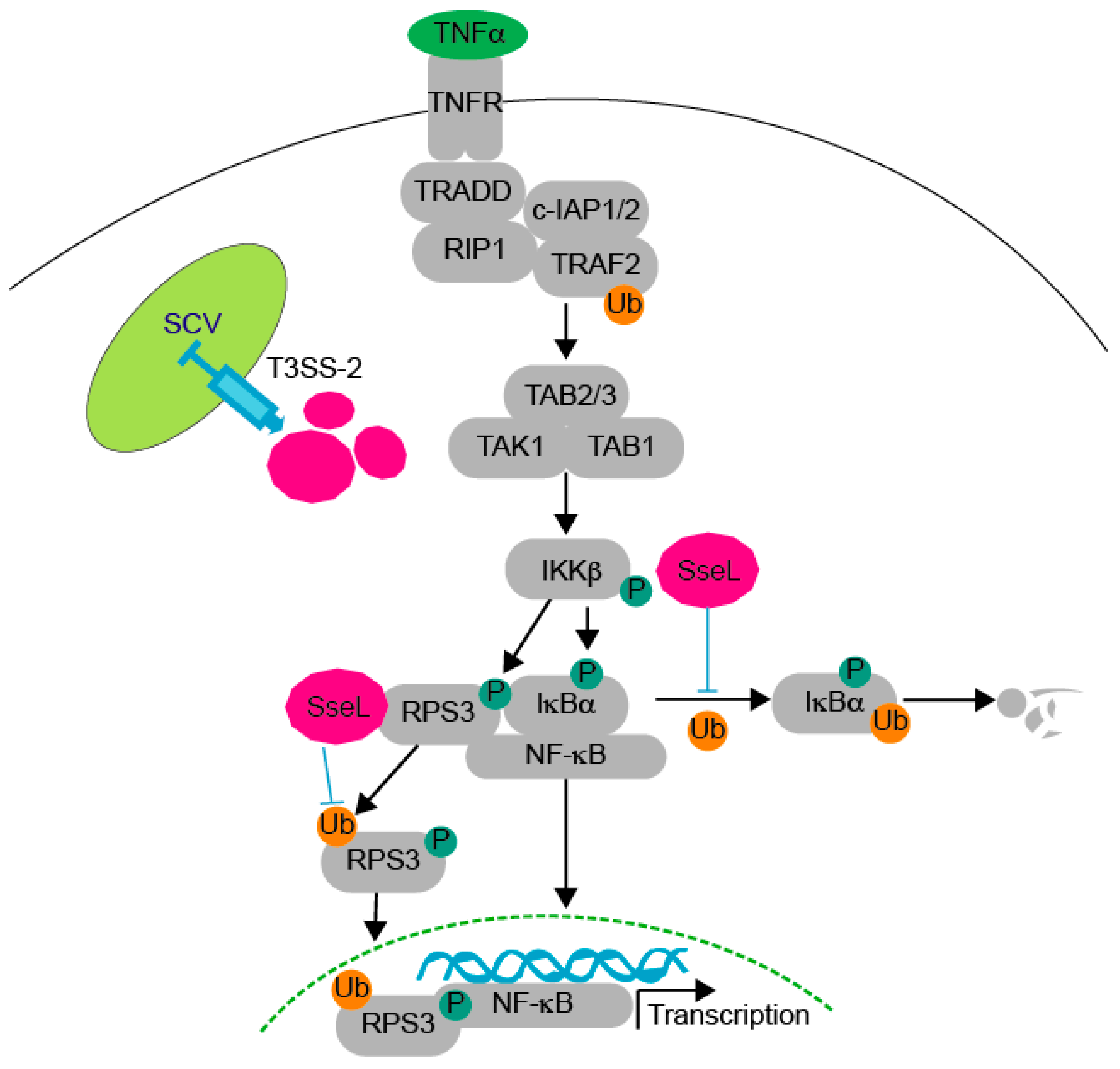
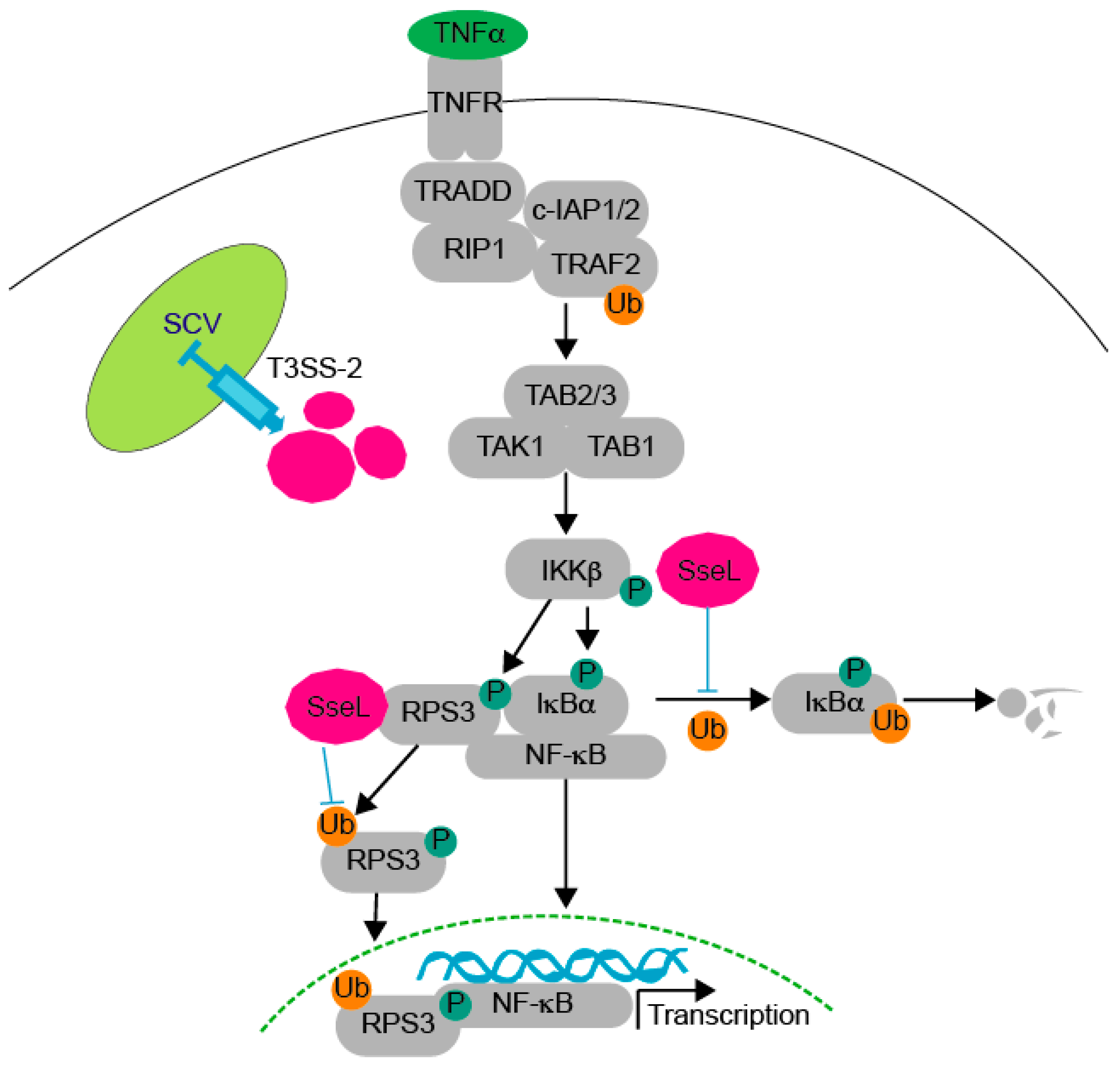
:1. Introduction
2. Results
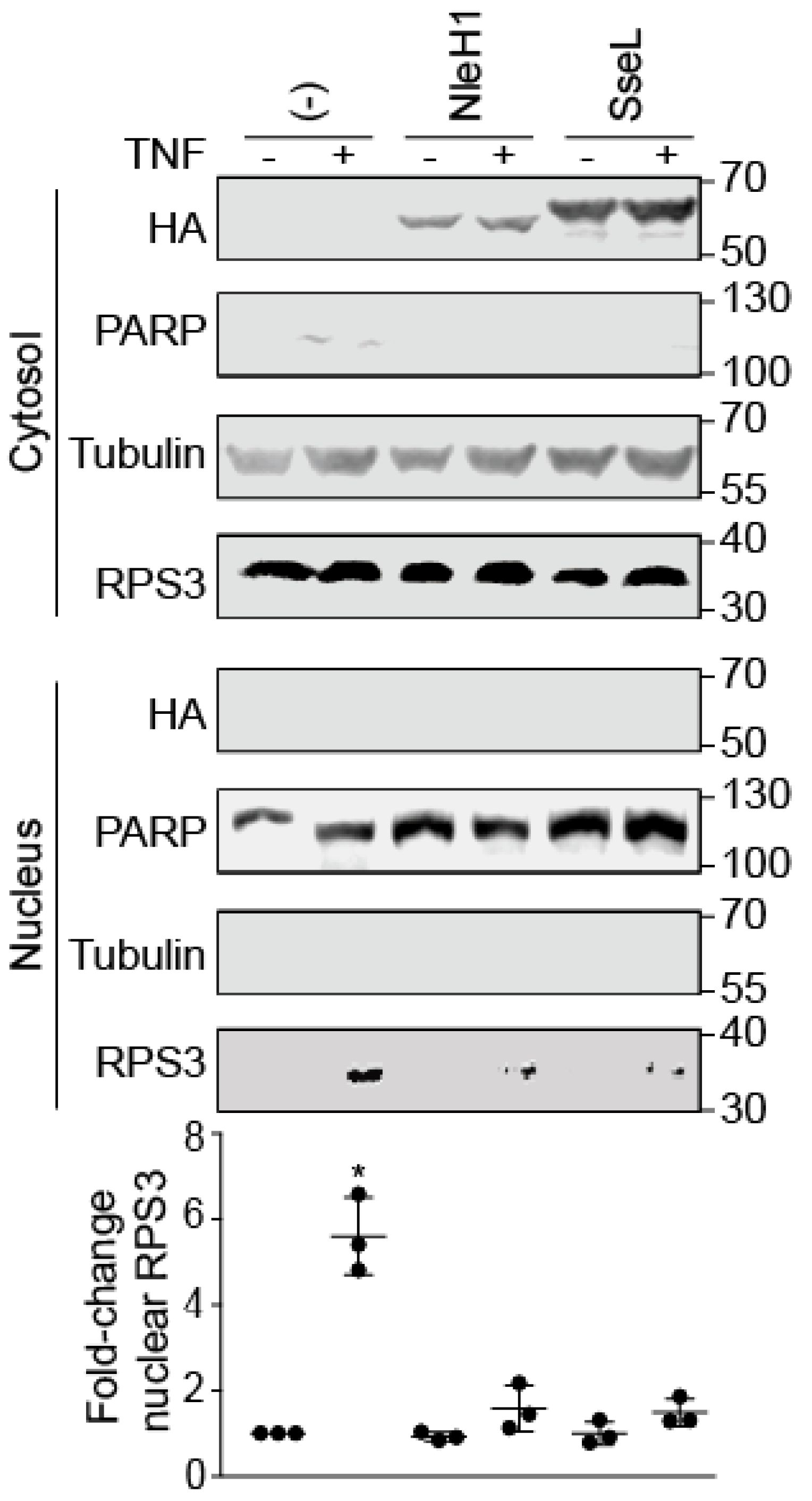
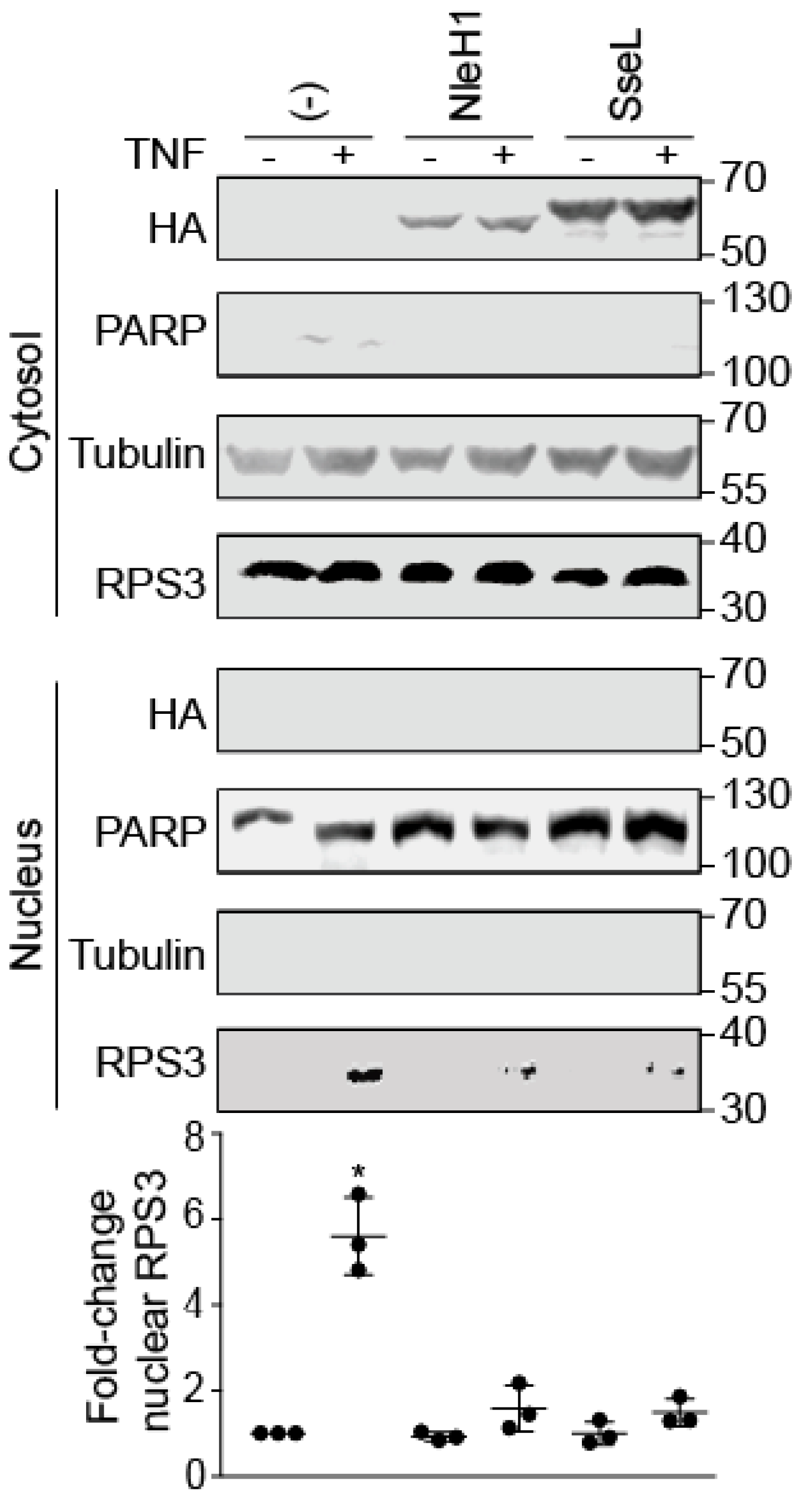
2.1. SseL Reduces the Nuclear Abundance of RPS3
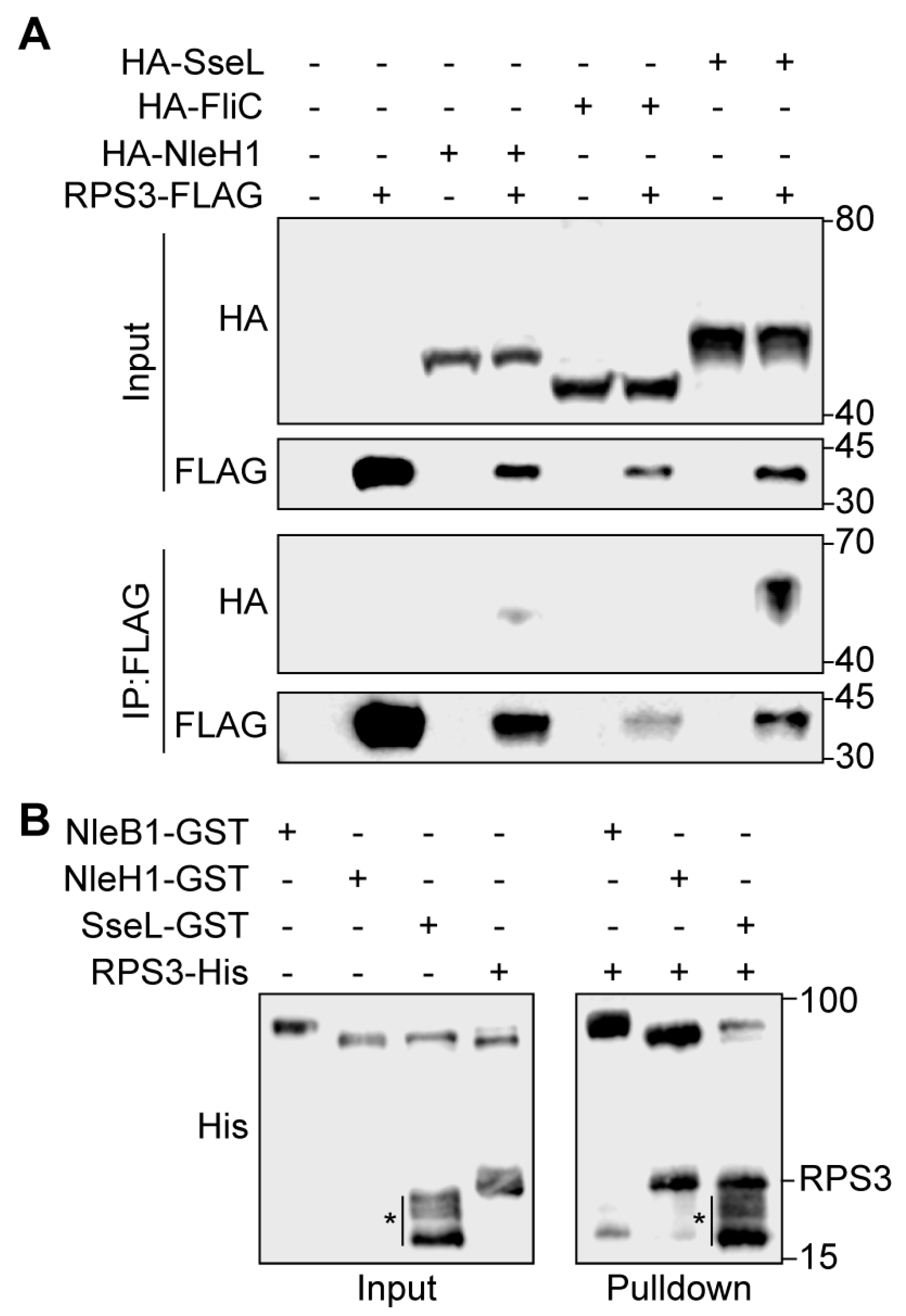
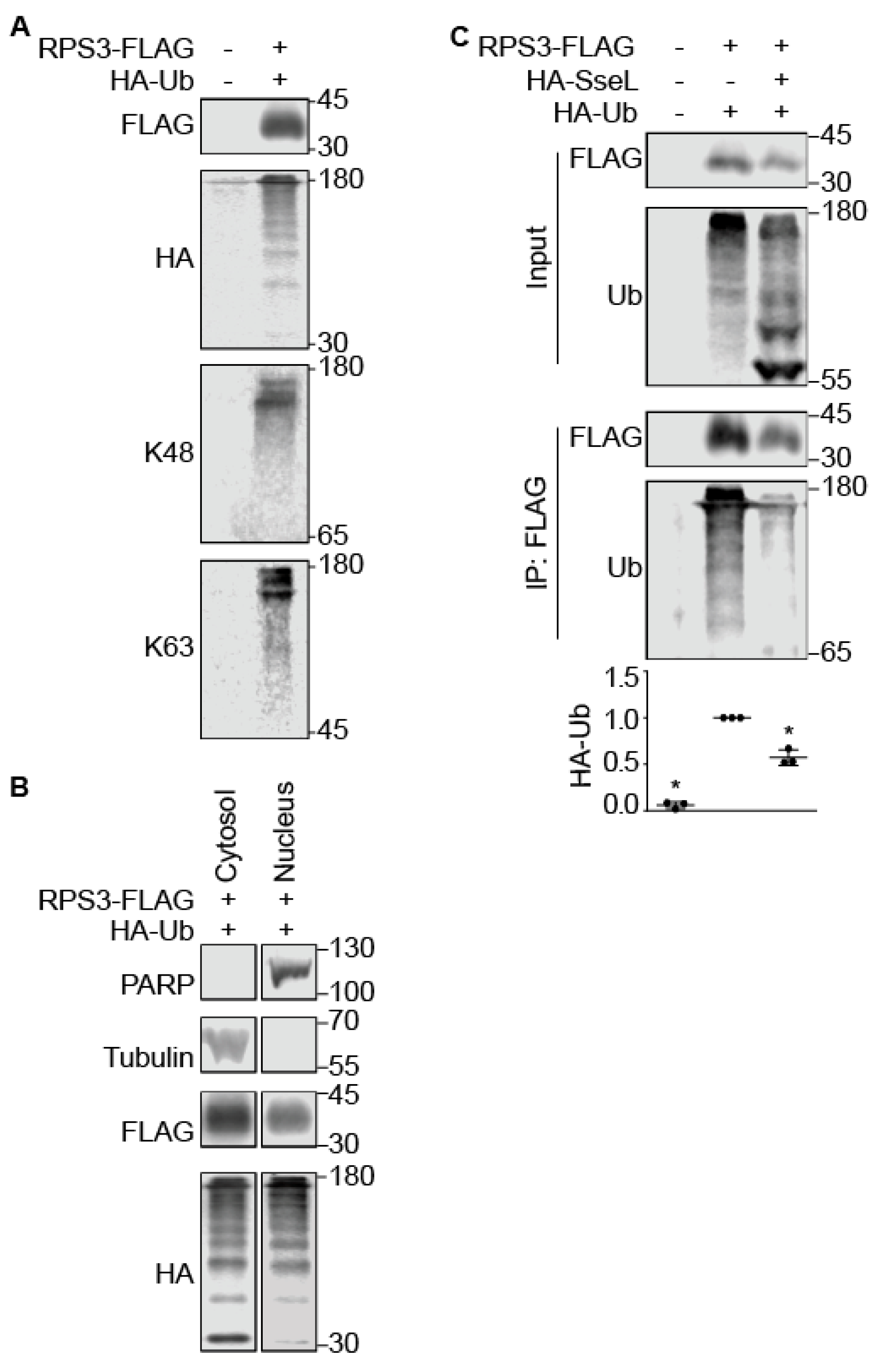
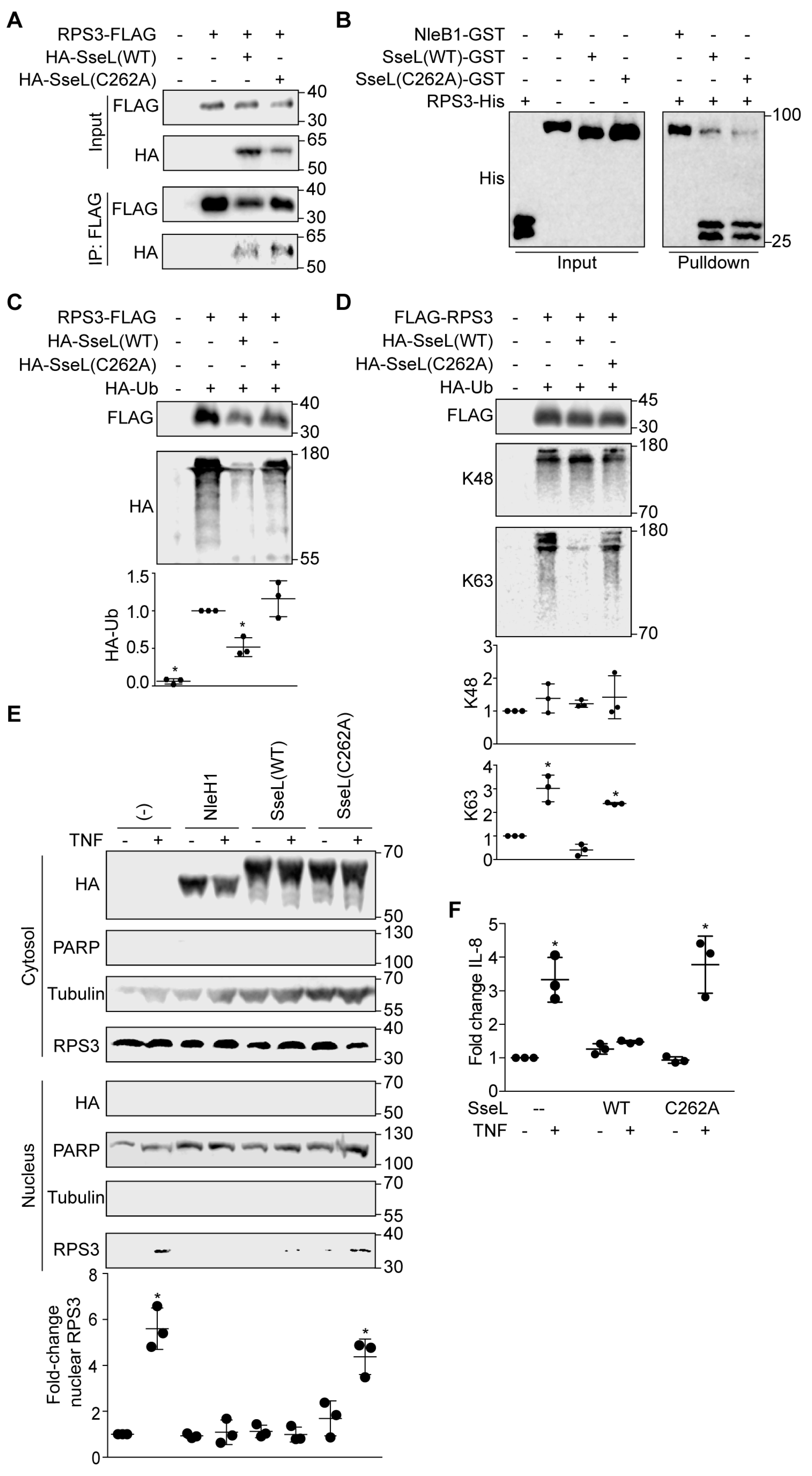
2.2. SseL Binds to RPS3
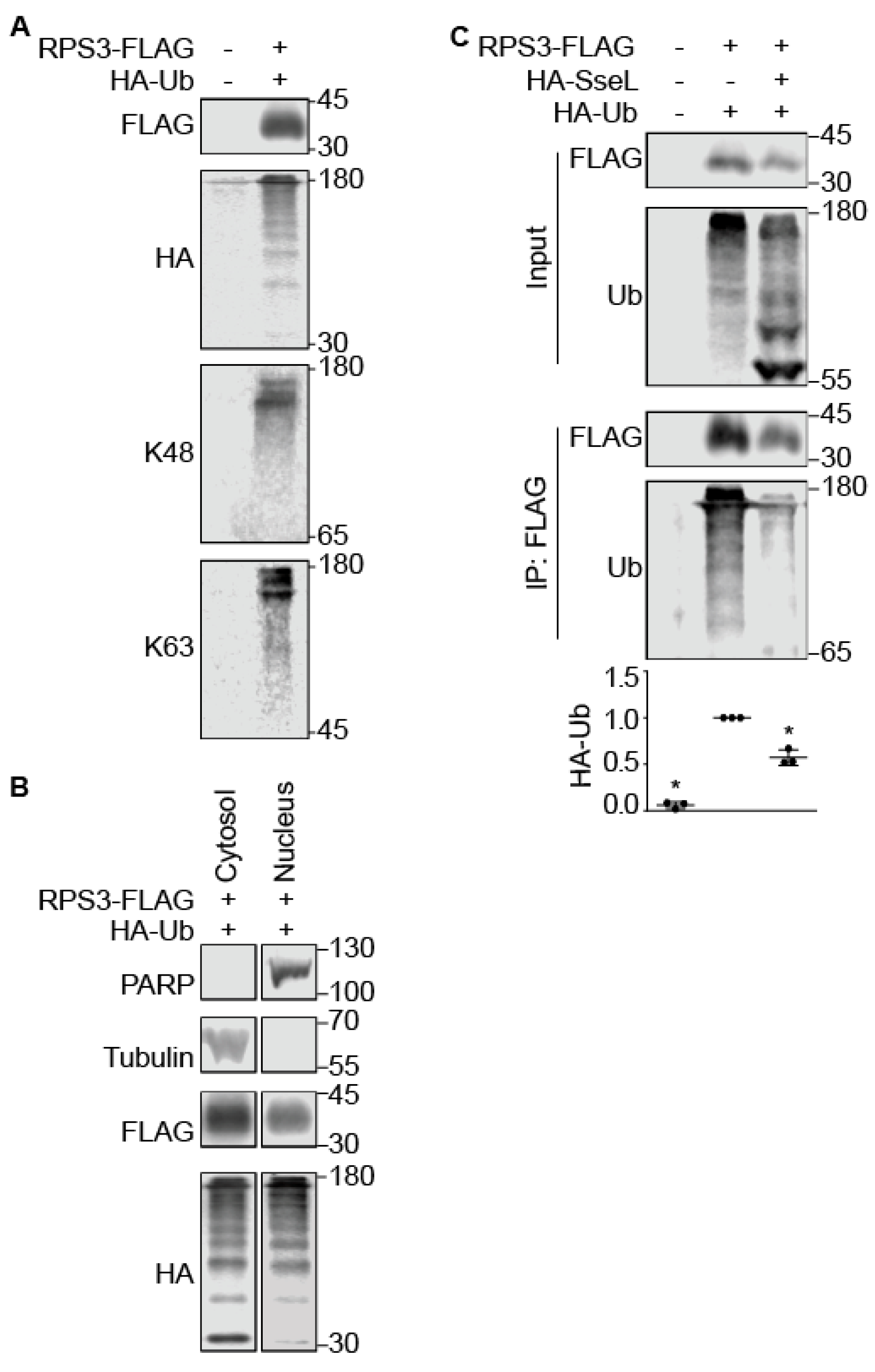
2.3. SseL Deubiquitinates RPS3
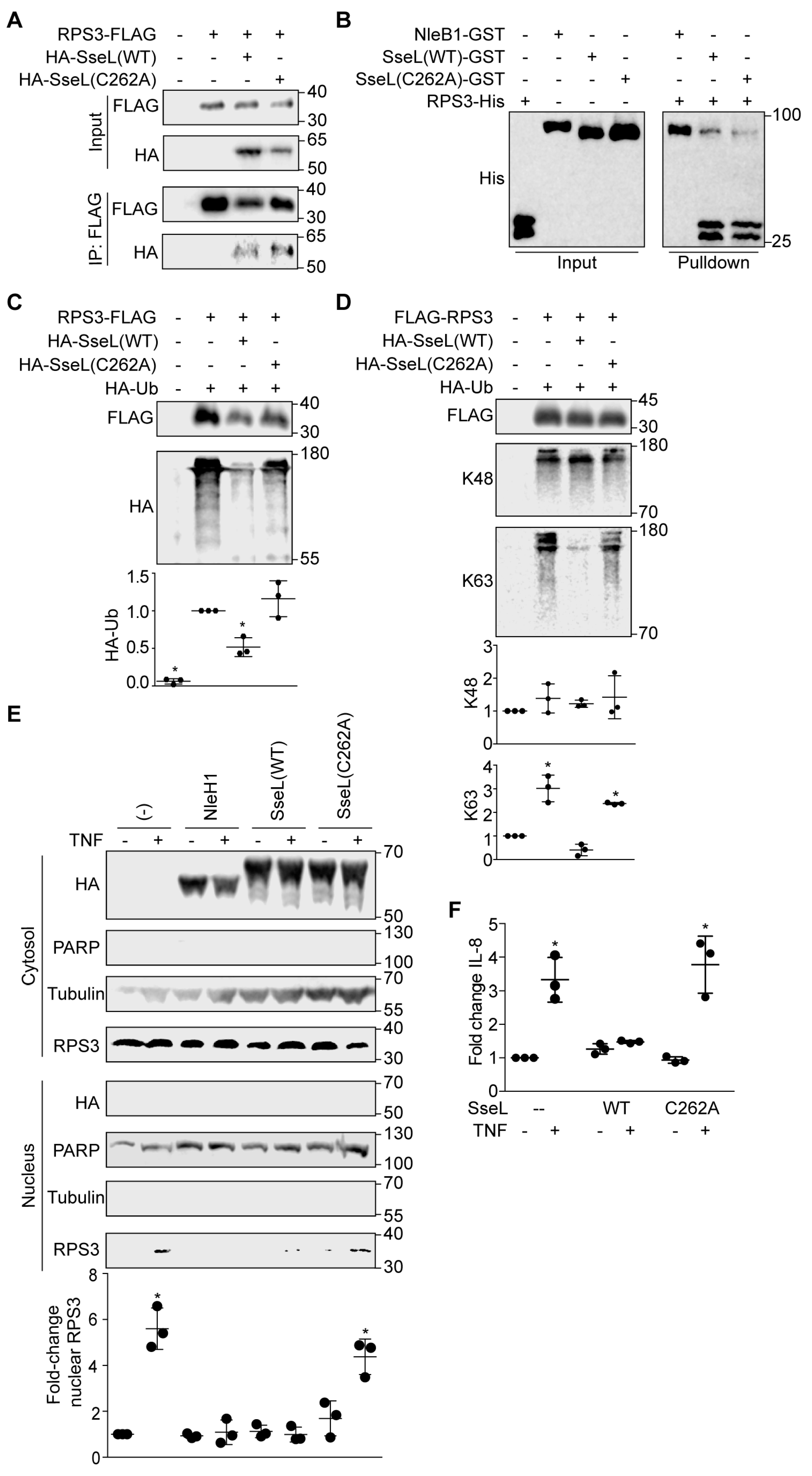
2.4. SseL DUB Activity Is Important to Inhibiting RPS3 Nuclear Translocation
3. Discussion
4. Materials and Methods
Author Contributions
Funding
Conflicts of Interest
References
- Galan, J.E.; Lara-Tejero, M.; Marlovits, T.C.; Wagner, S. Bacterial type iii secretion systems: Specialized nanomachines for protein delivery into target cells. Annu. Rev. Microbiol. 2014, 68, 415–438. [Google Scholar] [CrossRef] [PubMed]
- Newell, D.G.; La Ragione, R.M. Enterohaemorrhagic and other shiga toxin-producing escherichia coli (stec): Where are we now regarding diagnostics and control strategies? Transbound. Emerg. Dis. 2018, 65 (Suppl. 1), 49–71. [Google Scholar] [CrossRef]
- McDaniel, T.K.; Jarvis, K.G.; Donnenberg, M.S.; Kaper, J.B. A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc. Natl. Acad. Sci. USA 1995, 92, 1664–1668. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.C.; Galan, J.E. Manipulation of the host actin cytoskeleton by salmonella—All in the name of entry. Curr. Opin. Microbiol. 2005, 8, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Ochman, H.; Soncini, F.C.; Solomon, F.; Groisman, E.A. Identification of a pathogenicity island required for salmonella survival in host cells. Proc. Natl. Acad. Sci. USA 1996, 93, 7800–7804. [Google Scholar] [CrossRef] [PubMed]
- Le Negrate, G. Subversion of innate immune responses by bacterial hindrance of nf-kappab pathway. Cell. Microbiol. 2012, 14, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Hardwidge, P.R. Ribosomal protein s3: A multifunctional target of attaching/effacing bacterial pathogens. Front. Microbiol. 2011, 2, 137. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Youn, H.; Lee, S.; Kim, E.; Kim, D.; Sub Lee, J.; Lee, J.M.; Youn, B. Rnf138-mediated ubiquitination of rps3 is required for resistance of glioblastoma cells to radiation-induced apoptosis. Exp. Mol. Med. 2018, 50, e434. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Kwon, I.S.; Park, J.; Lee, K.H.; Ahn, Y.; Lee, C.; Kim, J.; Choi, S.Y.; Cho, S.W.; Ahn, J.Y. Ribosomal protein s3, a new substrate of akt, serves as a signal mediator between neuronal apoptosis and DNA repair. J. Biol. Chem. 2010, 285, 29457–29468. [Google Scholar] [CrossRef] [PubMed]
- Hegde, V.; Yadavilli, S.; Deutsch, W.A. Knockdown of ribosomal protein s3 protects human cells from genotoxic stress. DNA Repair 2007, 6, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.S.; Kim, H.D.; Kim, J. Pkcdelta-dependent functional switch of rps3 between translation and DNA repair. Biochim. Biophys. Acta 2009, 1793, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liao, W.J.; Liao, J.M.; Liao, P.; Lu, H. Ribosomal proteins: Functions beyond the ribosome. J. Mol. Cell Biol. 2015, 7, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Fregoso, O.I.; Das, S.; Akerman, M.; Krainer, A.R. Splicing-factor oncoprotein srsf1 stabilizes p53 via rpl5 and induces cellular senescence. Mol. Cell 2013, 50, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, B.; Sampath, P.; Seshadri, V.; Maitra, R.K.; DiCorleto, P.E.; Fox, P.L. Regulated release of l13a from the 60s ribosomal subunit as a mechanism of transcript-specific translational control. Cell 2003, 115, 187–198. [Google Scholar] [CrossRef]
- Anderson, S.J.; Lauritsen, J.P.; Hartman, M.G.; Foushee, A.M.; Lefebvre, J.M.; Shinton, S.A.; Gerhardt, B.; Hardy, R.R.; Oravecz, T.; Wiest, D.L. Ablation of ribosomal protein l22 selectively impairs alphabeta t cell development by activation of a p53-dependent checkpoint. Immunity 2007, 26, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Wan, F.; Anderson, D.E.; Barnitz, R.A.; Snow, A.; Bidere, N.; Zheng, L.; Hegde, V.; Lam, L.T.; Staudt, L.M.; Levens, D.; et al. Ribosomal protein s3: A kh domain subunit in nf-kappab complexes that mediates selective gene regulation. Cell 2007, 131, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Alkalay, I.; Yaron, A.; Hatzubai, A.; Orian, A.; Ciechanover, A.; Ben-Neriah, Y. Stimulation-dependent i kappa b alpha phosphorylation marks the nf-kappa b inhibitor for degradation via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA 1995, 92, 10599–10603. [Google Scholar] [CrossRef] [PubMed]
- Wan, F.; Weaver, A.; Gao, X.; Bern, M.; Hardwidge, P.R.; Lenardo, M.J. Ikkbeta phosphorylation regulates rps3 nuclear translocation and nf-kappab function during infection with escherichia coli strain o157:H7. Nat. Immunol. 2011, 12, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Wan, F.; Mateo, K.; Callegari, E.; Wang, D.; Deng, W.; Puente, J.; Li, F.; Chaussee, M.S.; Finlay, B.B.; et al. Bacterial effector binding to ribosomal protein s3 subverts nf-kappab function. PLoS Pathog. 2009, 5, e1000708. [Google Scholar] [CrossRef] [PubMed]
- Wier, E.M.; Neighoff, J.; Sun, X.; Fu, K.; Wan, F. Identification of an n-terminal truncation of the nf-kappab p65 subunit that specifically modulates ribosomal protein s3-dependent nf-kappab gene expression. J. Biol. Chem. 2012, 287, 43019–43029. [Google Scholar] [CrossRef] [PubMed]
- Le Negrate, G.; Faustin, B.; Welsh, K.; Loeffler, M.; Krajewska, M.; Hasegawa, P.; Mukherjee, S.; Orth, K.; Krajewski, S.; Godzik, A.; et al. Salmonella secreted factor l deubiquitinase of salmonella typhimurium inhibits nf-kappab, suppresses ikappabalpha ubiquitination and modulates innate immune responses. J. Immunol. 2008, 180, 5045–5056. [Google Scholar] [CrossRef] [PubMed]
- Rytkonen, A.; Poh, J.; Garmendia, J.; Boyle, C.; Thompson, A.; Liu, M.; Freemont, P.; Hinton, J.C.; Holden, D.W. Ssel, a salmonella deubiquitinase required for macrophage killing and virulence. Proc. Natl. Acad. Sci. USA 2007, 104, 3502–3507. [Google Scholar] [CrossRef] [PubMed]
- El Qaidi, S.; Chen, K.; Halim, A.; Siukstaite, L.; Rueter, C.; Hurtado-Guerrero, R.; Clausen, H.; Hardwidge, P.R. Nleb/ssek effectors from citrobacter rodentium, escherichia coli, and salmonella enterica display distinct differences in host substrate specificity. J. Biol. Chem. 2017, 292, 11423–11430. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Kim, H.D.; Yang, H.W.; Kim, H.J.; Jang, C.Y.; Kim, J. Modulating cellular balance of rps3 mono-ubiquitination by both hel2 e3 ligase and ubp3 deubiquitinase regulates protein quality control. Exp. Mol. Med. 2017, 49, e390. [Google Scholar] [CrossRef] [PubMed]
- Simms, C.L.; Yan, L.L.; Zaher, H.S. Ribosome collision is critical for quality control during no-go decay. Mol. Cell 2017, 68, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Wan, F.; Lenardo, M.J. Specification of DNA binding activity of nf-kappab proteins. Cold Spring Harb. Perspect. Biol. 2009, 1, a000067. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Saide, A.; Cagliani, R.; Cantile, M.; Botti, G.; Russo, G. Rpl3 promotes the apoptosis of p53 mutated lung cancer cells by down-regulating cbs and nfkappab upon 5-fu treatment. Sci. Rep. 2016, 6, 38369. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Paul, B.D.; Gadalla, M.M.; Mustafa, A.K.; Sen, T.; Xu, R.; Kim, S.; Snyder, S.H. Hydrogen sulfide-linked sulfhydration of nf-kappab mediates its antiapoptotic actions. Mol. Cell 2012, 45, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Danielsen, J.M.; Sylvestersen, K.B.; Bekker-Jensen, S.; Szklarczyk, D.; Poulsen, J.W.; Horn, H.; Jensen, L.J.; Mailand, N.; Nielsen, M.L. Mass spectrometric analysis of lysine ubiquitylation reveals promiscuity at site level. Mol. Cell. Proteom. 2011, 10, M110.003590. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Chan, D.W.; Jung, S.Y.; Malovannaya, A.; Wang, Y.; Qin, J. A data set of human endogenous protein ubiquitination sites. Mol. Cell. Proteom. 2011, 10, M110.002089. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.A.; Beli, P.; Weinert, B.T.; Nielsen, M.L.; Cox, J.; Mann, M.; Choudhary, C. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol. Cell. Proteom. 2011, 10, M111.013284. [Google Scholar] [CrossRef] [PubMed]
- Higgins, R.; Gendron, J.M.; Rising, L.; Mak, R.; Webb, K.; Kaiser, S.E.; Zuzow, N.; Riviere, P.; Yang, B.; Fenech, E.; et al. The unfolded protein response triggers site-specific regulatory ubiquitylation of 40s ribosomal proteins. Mol. Cell 2015, 59, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Sundaramoorthy, E.; Leonard, M.; Mak, R.; Liao, J.; Fulzele, A.; Bennett, E.J. Znf598 and rack1 regulate mammalian ribosome-associated quality control function by mediating regulatory 40s ribosomal ubiquitylation. Mol. Cell 2017, 65, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Yadavilli, S.; Mayo, L.D.; Higgins, M.; Lain, S.; Hegde, V.; Deutsch, W.A. Ribosomal protein s3: A multi-functional protein that interacts with both p53 and mdm2 through its kh domain. DNA Repair 2009, 8, 1215–1224. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.S.; Jang, C.Y.; Kim, H.D.; Lee, J.Y.; Ahn, B.Y.; Kim, J. Interaction of hsp90 with ribosomal proteins protects from ubiquitination and proteasome-dependent degradation. Mol. Biol. Cell 2006, 17, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Beverly, L.J.; Lockwood, W.W.; Shah, P.P.; Erdjument-Bromage, H.; Varmus, H. Ubiquitination, localization, and stability of an anti-apoptotic bcl2-like protein, bcl2l10/bclb, are regulated by ubiquilin1. Proc. Natl. Acad. Sci. USA 2012, 109, E119–E126. [Google Scholar] [CrossRef] [PubMed]
- Gregory, R.C.; Taniguchi, T.; D’Andrea, A.D. Regulation of the fanconi anemia pathway by monoubiquitination. Semin. Cancer Biol. 2003, 13, 77–82. [Google Scholar] [CrossRef]
- Wang, P.Y.; Chang, K.T.; Lin, Y.M.; Kuo, T.Y.; Wang, G.S. Ubiquitination of mbnl1 is required for its cytoplasmic localization and function in promoting neurite outgrowth. Cell Rep. 2018, 22, 2294–2306. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Geisbrecht, B.V.; Rueter, C.; Hardwidge, P.R. Enterotoxigenic escherichia coli flagellin inhibits tnf-induced nf-kappab activation in intestinal epithelial cells. Pathogens 2017, 6, 18. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain/Plasmid | Description | Source |
|---|---|---|
| Strain | ||
| E. coli BL21(DE3) | E. coli F−ompT hsdSB (rB−_m B−) gal dcm (DE3) | Novagen |
| BL21(DE3)/NleH1-pET42a | GST-EHEC NleH1 | [19] |
| BL21(DE3)/SseL-pET42a | GST-S. Typhimurium SseL | This study |
| BL21(DE3)/RPS3-pET28a | His-RPS3 | This study |
| BL21(DE3)NleB1-pET42a | GST-EHEC NleB1 | [23] |
| BL21(DE3)/SseL(C262A)-pET42a | GST-S. Typhimurium SseL(C262A) | This study |
| Plasmid | ||
| HA | HA fusion expression | Clontech |
| NleH1-HA | HA fused to E. coli EDL933 NleH1 | [19] |
| SseL-HA | HA fused to S. Typhimurium SseL | This study |
| SseL(C262A)-HA | HA fused to S. Typhimurium SseL(C262A) | This study |
| FliC-HA | HA fused to ETEC FliC | [39] |
| 3× FLAG | FLAG expression | Sigma |
| 3× FLAG-RPS3 | FLAG-RPS3 | [16] |
| pET42a | Bacterial GST fusion expression | Novagen |
| NleH1-pET42a | GST-EHEC NleH1 | [19] |
| NleB1-pET42a | GST-EHEC NleB1 | [23] |
| SseL-pET42a | GST-S. Typhimurium SseL | This study |
| SseL(C262A)-pET42a | GST-S. Typhimurium SseL(C262A) | This study |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, M.; El Qaidi, S.; Hardwidge, P.R. SseL Deubiquitinates RPS3 to Inhibit Its Nuclear Translocation. Pathogens 2018, 7, 86. https://doi.org/10.3390/pathogens7040086
Wu M, El Qaidi S, Hardwidge PR. SseL Deubiquitinates RPS3 to Inhibit Its Nuclear Translocation. Pathogens. 2018; 7(4):86. https://doi.org/10.3390/pathogens7040086
Chicago/Turabian StyleWu, Miaomiao, Samir El Qaidi, and Philip R. Hardwidge. 2018. "SseL Deubiquitinates RPS3 to Inhibit Its Nuclear Translocation" Pathogens 7, no. 4: 86. https://doi.org/10.3390/pathogens7040086
APA StyleWu, M., El Qaidi, S., & Hardwidge, P. R. (2018). SseL Deubiquitinates RPS3 to Inhibit Its Nuclear Translocation. Pathogens, 7(4), 86. https://doi.org/10.3390/pathogens7040086