Listeria Monocytogenes: A Model Pathogen Continues to Refine Our Knowledge of the CD8 T Cell Response

{kind=link}
Abstract
1. Introduction
2. Listeria Monocytogenes (Lm) Acquisition and Presentation by Dendritic Cells (DC)
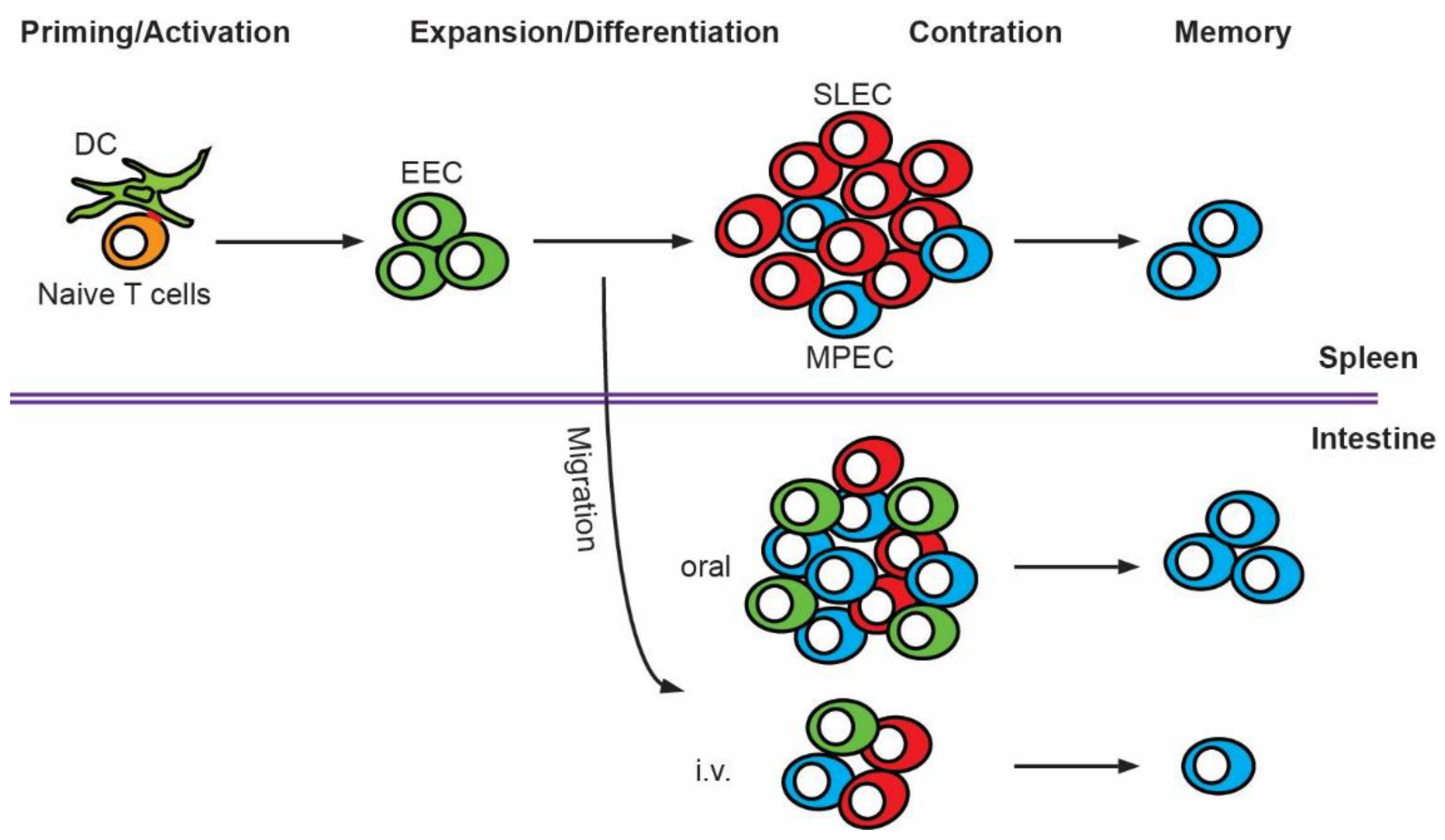
3. T Cell Priming and Activation
4. T Cell Expansion, Differentiation and Contraction
5. T Cell Memory Formation
6. CD4 T Cell Help
7. CD8 T Cell-Mediated Protective Immunity against Lm Infection
8. Non-Classical H2-M3-Restricited CD8 T Cell Response
9. Lm as a Vaccine Vector for Cancer Immunotherapy
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Scallan, E.; Hoekstra, R.M.; Angulo, F.J.; Tauxe, R.V.; Widdowson, M.; Roy, S.L.; Jones, J.L.; Griffin, P.M. Foodborne Illness Acquired in the United States-Major Pathogens. Emerg. Infect. Dis. 2011, 17, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.; Batz, M.B.; Morris, J.G., Jr. Annual cost of illness and quality-adjusted life year losses in the United States due to 14 foodborne pathogens. J. Food Prot. 2012, 75, 1292–1302. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.; Maculloch, B.; Batz, M. Economic Burden of Major Foodborne Illnesses Acquired in the United States. EIB-140. May 2015; U.S. Department of Agriculture, Economic Research Service: Washington, DC, USA, 2015.
- Freitag, N.E.; Port, G.C.; Miner, M.D. Listeria monocytogenes—From saprophyte to intracellular pathogen. Nat. Rev. Microbiol. 2009, 7, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Lecuit, M.; Dramsi, S.; Gottardi, C.; Fedor-Chaiken, M.; Gumbiner, B.; Cossart, P. A single amino acid in e-cadherin responsible for host specificity towards the human pathogen Listeria monocytogenes. EMBO J. 1999, 18, 3956–3963. [Google Scholar] [CrossRef] [PubMed]
- Lecuit, M.; Vandormael-Pournin, S.; Lefort, J.; Huerre, M.; Gounon, P.; Dupuy, C.; Babinet, C.; Cossart, P. A transgenic model for listeriosis: Role of internalin in crossing the intestinal barrier. Science 2001, 292, 1722–1725. [Google Scholar] [CrossRef] [PubMed]
- Disson, O.; Grayo, S.; Huillet, E.; Nikitas, G.; Langa-Vives, F.; Dussurget, O.; Ragon, M.; Le Monnier, A.; Babinet, C.; Cossart, P.; et al. Conjugated action of two species-specific invasion proteins for fetoplacental listeriosis. Nature 2008, 455, 1114–1118. [Google Scholar] [CrossRef] [PubMed]
- Wollert, T.; Pasche, B.; Rochon, M.; Deppenmeier, S.; van den Heuvel, J.; Gruber, A.D.; Heinz, D.W.; Lengeling, A.; Schubert, W.D. Extending the host range of Listeria monocytogenes by rational protein design. Cell 2007, 129, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, B.S.; Romagnoli, P.A.; Pham, Q.M.; Fu, H.H.; Alonzo, F., 3rd; Schubert, W.D.; Freitag, N.E.; Lefrancois, L. Gammadelta T cells exhibit multifunctional and protective memory in intestinal tissues. Immunity 2013, 39, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Bou Ghanem, E.N.; Jones, G.S.; Myers-Morales, T.; Patil, P.D.; Hidayatullah, A.N.; D’Orazio, S.E. Inla promotes dissemination of Listeria monocytogenes to the mesenteric lymph nodes during food borne infection of mice. PLoS Pathog. 2012, 8, e1003015. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.J.; Liang, H.E.; Reizis, B.; Locksley, R.M. Regulation of hierarchical clustering and activation of innate immune cells by dendritic cells. Immunity 2008, 29, 819–833. [Google Scholar] [CrossRef] [PubMed]
- Emmerling, P.; Finger, H.; Bockemuhl, J. Listeria monocytogenes infection in nude mice. Infect. Immun. 1975, 12, 437–439. [Google Scholar] [PubMed]
- Ladel, C.H.; Flesch, I.E.; Arnoldi, J.; Kaufmann, S.H. Studies with mhc-deficient knock-out mice reveal impact of both mhc i- and mhc ii-dependent T cell responses on Listeria monocytogenes infection. J. Immunol. 1994, 153, 3116–3122. [Google Scholar] [PubMed]
- Bhardwaj, V.; Kanagawa, O.; Swanson, P.E.; Unanue, E.R. Chronic listeria infection in SCID mice: Requirements for the carrier state and the dual role of T cells in transferring protection or suppression. J. Immunol. 1998, 160, 376–384. [Google Scholar] [PubMed]
- Romagnoli, P.A.; Fu, H.H.; Qiu, Z.; Khairallah, C.; Pham, Q.M.; Puddington, L.; Khanna, K.M.; Lefrancois, L.; Sheridan, B.S. Differentiation of distinct long-lived memory CD4 T cells in intestinal tissues after oral Listeria monocytogenes infection. Mucosal Immunol. 2017, 10, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Condotta, S.A.; Richer, M.J.; Badovinac, V.P.; Harty, J.T. Probing CD8 T cell responses with Listeria monocytogenes infection. Adv. Immunol. 2012, 113, 51–80. [Google Scholar] [PubMed]
- Khan, S.H.; Badovinac, V.P. Listeria monocytogenes: A model pathogen to study antigen-specific memory CD8 T cell responses. Semin. Immunopathol. 2015, 37, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Aichele, P.; Zinke, J.; Grode, L.; Schwendener, R.A.; Kaufmann, S.H.; Seiler, P. Macrophages of the splenic marginal zone are essential for trapping of blood-borne particulate antigen but dispensable for induction of specific T cell responses. J. Immunol. 2003, 171, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Aoshi, T.; Carrero, J.A.; Konjufca, V.; Koide, Y.; Unanue, E.R.; Miller, M.J. The cellular niche of Listeria monocytogenes infection changes rapidly in the spleen. Eur. J. Immunol. 2009, 39, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Perez, O.A.; Yeung, S.T.; Vera-Licona, P.; Romagnoli, P.A.; Samji, T.; Ural, B.B.; Maher, L.; Tanaka, M.; Khanna, K.M. Cd169+ macrophages orchestrate innate immune responses by regulating bacterial localization in the spleen. Sci. Immunol. 2017, 2, eaah5520. [Google Scholar] [CrossRef] [PubMed]
- Conlan, J.W. Early pathogenesis of Listeria monocytogenes infection in the mouse spleen. J. Med. Microbiol. 1996, 44, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Aoshi, T.; Zinselmeyer, B.H.; Konjufca, V.; Lynch, J.N.; Zhang, X.; Koide, Y.; Miller, M.J. Bacterial entry to the splenic white pulp initiates antigen presentation to CD8+ T cells. Immunity 2008, 29, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Neuenhahn, M.; Kerksiek, K.M.; Nauerth, M.; Suhre, M.H.; Schiemann, M.; Gebhardt, F.E.; Stemberger, C.; Panthel, K.; Schroder, S.; Chakraborty, T.; et al. Cd8α+ dendritic cells are required for efficient entry of Listeria monocytogenes into the spleen. Immunity 2006, 25, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Edelson, B.T.; Bradstreet, T.R.; Hildner, K.; Carrero, J.A.; Frederick, K.E.; Kc, W.; Belizaire, R.; Aoshi, T.; Schreiber, R.D.; Miller, M.J.; et al. Cd8α+ dendritic cells are an obligate cellular entry point for productive infection by Listeria monocytogenes. Immunity 2011, 35, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Verschoor, A.; Neuenhahn, M.; Navarini, A.A.; Graef, P.; Plaumann, A.; Seidlmeier, A.; Nieswandt, B.; Massberg, S.; Zinkernagel, R.M.; Hengartner, H.; et al. A platelet-mediated system for shuttling blood-borne bacteria to CD8α+ dendritic cells depends on glycoprotein gpib and complement C3. Nat. Immunol. 2011, 12, 1194–1201. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, D.; Sadikovic, A.; Vanloubbeeck, Y.; Brockstedt, D.; Fong, L. Interplay between CD8α+ dendritic cells and monocytes in response to Listeria monocytogenes infection attenuates T cell responses. PLoS ONE 2011, 6, e19376. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.E.; Schmidt, R.L.; McDermott, D.S.; Lenz, L.L. A Batf3/Nlrp3/IL-18 Axis Promotes Natural Killer Cell IL-10 Production during Listeria monocytogenes infection. Cell Rep. 2018, 23, 2582–2594. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.E.; Filak, H.C.; Guthrie, B.S.; Schmidt, R.L.; Jamieson, A.; Merkel, P.; Knight, V.; Cole, C.M.; Raulet, D.H.; Lenz, L.L. Bacterial manipulation of nk cell regulatory activity increases susceptibility to Listeria monocytogenes infection. PLoS Pathog. 2016, 12, e1005708. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.S.; Bussell, K.M.; Myers-Morales, T.; Fieldhouse, A.M.; Bou Ghanem, E.N.; D’Orazio, S.E. Intracellular Listeria monocytogenes comprises a minimal but vital fraction of the intestinal burden following foodborne infection. Infect. Immun. 2015, 83, 3146–3156. [Google Scholar] [CrossRef] [PubMed]
- Niess, J.H.; Brand, S.; Gu, X.; Landsman, L.; Jung, S.; McCormick, B.A.; Vyas, J.M.; Boes, M.; Ploegh, H.L.; Fox, J.G.; et al. Cx3cr1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 2005, 307, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Diehl, G.E.; Longman, R.S.; Zhang, J.X.; Breart, B.; Galan, C.; Cuesta, A.; Schwab, S.R.; Littman, D.R. Microbiota restricts trafficking of bacteria to mesenteric lymph nodes by CX3CR1hi cells. Nature 2013, 494, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Farache, J.; Koren, I.; Milo, I.; Gurevich, I.; Kim, K.W.; Zigmond, E.; Furtado, G.C.; Lira, S.A.; Shakhar, G. Luminal bacteria recruit CD103+ dendritic cells into the intestinal epithelium to sample bacterial antigens for presentation. Immunity 2013, 38, 581–595. [Google Scholar] [CrossRef] [PubMed]
- Schulz, O.; Jaensson, E.; Persson, E.K.; Liu, X.; Worbs, T.; Agace, W.W.; Pabst, O. Intestinal CD103+, but not CX3CR1+, antigen sampling cells migrate in lymph and serve classical dendritic cell functions. J. Exp. Med. 2009, 206, 3101–3114. [Google Scholar] [CrossRef] [PubMed]
- McDole, J.R.; Wheeler, L.W.; McDonald, K.G.; Wang, B.; Konjufca, V.; Knoop, K.A.; Newberry, R.D.; Miller, M.J. GobleT cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature 2012, 483, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Mazzini, E.; Massimiliano, L.; Penna, G.; Rescigno, M. Oral tolerance can be established via gap junction transfer of fed antigens from CX3CR1+ macrophages to CD103+ dendritic cells. Immunity 2014, 40, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Nikitas, G.; Deschamps, C.; Disson, O.; Niault, T.; Cossart, P.; Lecuit, M. Transcytosis of Listeria monocytogenes across the intestinal barrier upon specific targeting of gobleT cell accessible e-cadherin. J. Exp. Med. 2011, 208, 2263–2277. [Google Scholar] [CrossRef] [PubMed]
- Johansson-Lindbom, B.; Svensson, M.; Pabst, O.; Palmqvist, C.; Marquez, G.; Forster, R.; Agace, W.W. Functional specialization of gut CD103+ dendritic cells in the regulation of tissue-selective T cell homing. J. Exp. Med. 2005, 202, 1063–1073. [Google Scholar] [CrossRef] [PubMed]
- Persson, E.K.; Scott, C.L.; Mowat, A.M.; Agace, W.W. Dendritic cell subsets in the intestinal lamina propria: Ontogeny and function. Eur. J. Immunol. 2013, 43, 3098–3107. [Google Scholar] [CrossRef] [PubMed]
- Edelson, B.T.; Kc, W.; Juang, R.; Kohyama, M.; Benoit, L.A.; Klekotka, P.A.; Moon, C.; Albring, J.C.; Ise, W.; Michael, D.G.; et al. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8α+ conventional dendritic cells. J. Exp. Med. 2010, 207, 823–836. [Google Scholar] [CrossRef] [PubMed]
- Persson, E.K.; Uronen-Hansson, H.; Semmrich, M.; Rivollier, A.; Hagerbrand, K.; Marsal, J.; Gudjonsson, S.; Hakansson, U.; Reizis, B.; Kotarsky, K.; et al. IRF4 transcription-factor-dependent CD103+CD11b+ dendritic cells drive mucosal T helper 17 cell differentiation. Immunity 2013, 38, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, K.; Karuppuchamy, T.; Takemura, N.; Shimohigoshi, M.; Machida, T.; Haseda, Y.; Aoshi, T.; Ishii, K.J.; Akira, S.; Uematsu, S. A new subset of CD103+CD8α+ dendritic cells in the small intestine expresses TLR3, TLR7, and TLR9 and induces TH1 response and CTL activity. J. Immunol. 2011, 186, 6287–6295. [Google Scholar] [CrossRef] [PubMed]
- Pepper, M.; Linehan, J.L.; Pagan, A.J.; Zell, T.; Dileepan, T.; Cleary, P.P.; Jenkins, M.K. Different routes of bacterial infection induce long-lived th1 memory cells and short-lived th17 cells. Nat. Immunol. 2010, 11, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Mempel, T.R.; Henrickson, S.E.; Von Andrian, U.H. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature 2004, 427, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Khanna, K.M.; McNamara, J.T.; Lefrancois, L. In situ imaging of the endogenous CD8 T cell response to infection. Science 2007, 318, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Busch, D.H.; Pilip, I.M.; Vijh, S.; Pamer, E.G. Coordinate regulation of complex T cell populations responding to bacterial infection. Immunity 1998, 8, 353–362. [Google Scholar] [CrossRef]
- Obar, J.J.; Jellison, E.R.; Sheridan, B.S.; Blair, D.A.; Pham, Q.M.; Zickovich, J.M.; Lefrancois, L. Pathogen-induced inflammatory environment controls effector and memory CD8+ T cell differentiation. J. Immunol. 2011, 187, 4967–4978. [Google Scholar] [CrossRef] [PubMed]
- Pope, C.; Kim, S.K.; Marzo, A.; Masopust, D.; Williams, K.; Jiang, J.; Shen, H.; Lefrancois, L. Organ-specific regulation of the CD8 T cell response to Listeria monocytogenes infection. J. Immunol. 2001, 166, 3402–3409. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, B.S.; Pham, Q.M.; Lee, Y.T.; Cauley, L.S.; Puddington, L.; Lefrancois, L. Oral infection drives a distinct population of intestinal resident memory CD8+ T cells with enhanced protective function. Immunity 2014, 40, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Lauvau, G.; Vijh, S.; Kong, P.; Horng, T.; Kerksiek, K.; Serbina, N.; Tuma, R.A.; Pamer, E.G. Priming of memory but not effector CD8 T cells by a killed bacterial vaccine. Science 2001, 294, 1735–1739. [Google Scholar] [CrossRef] [PubMed]
- Khanna, K.M.; Blair, D.A.; Vella, A.T.; McSorley, S.J.; Datta, S.K.; Lefrancois, L. T cell and apc dynamics in situ control the outcome of vaccination. J. Immunol. 2010, 185, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Von Koenig, C.H.; Finger, H.; Hof, H. Failure of killed Listeria monocytogenes vaccine to produce protective immunity. Nature 1982, 297, 233–234. [Google Scholar] [CrossRef] [PubMed]
- Sashinami, H.; Nakane, A.; Iwakura, Y.; Sasaki, M. Effective induction of acquired resistance to Listeria monocytogenes by immunizing mice with in vivo-infected dendritic cells. Infect. Immun. 2003, 71, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.K.; Okamoto, S.; Hayashi, T.; Shin, S.S.; Mihajlov, I.; Fermin, A.; Guiney, D.G.; Fierer, J.; Raz, E. Vaccination with irradiated listeria induces protective T cell immunity. Immunity 2006, 25, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Zhang, D.; Palliser, D.; Zhu, P.; Cai, S.; Schlesinger, A.; Maliszewski, L.; Lieberman, J. Listeria-infected myeloid dendritic cells produce ifn-beta, priming T cell activation. J. Immunol. 2005, 175, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Muraille, E.; Giannino, R.; Guirnalda, P.; Leiner, I.; Jung, S.; Pamer, E.G.; Lauvau, G. Distinct in vivo dendritic cell activation by live versus killed Listeria monocytogenes. Eur. J. Immunol. 2005, 35, 1463–1471. [Google Scholar] [CrossRef] [PubMed]
- Mittrucker, H.W.; Kursar, M.; Kohler, A.; Hurwitz, R.; Kaufmann, S.H. Role of CD28 for the generation and expansion of antigen-specific CD8+ t lymphocytes during infection with Listeria monocytogenes. J. Immunol. 2001, 167, 5620–5627. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.H.; Sougawa, N.; Tanaka, T.; Hirata, T.; Hiroi, T.; Tohya, K.; Guo, Z.; Umemoto, E.; Ebisuno, Y.; Yang, B.G.; et al. CCR7 is critically important for migration of dendritic cells in intestinal lamina propria to mesenteric lymph nodes. J. Immunol. 2006, 176, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Blattman, J.N.; Antia, R.; Sourdive, D.J.; Wang, X.; Kaech, S.M.; Murali-Krishna, K.; Altman, J.D.; Ahmed, R. Estimating the precursor frequency of naive antigen-specific CD8 T cells. J. Exp. Med. 2002, 195, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Obar, J.J.; Khanna, K.M.; Lefrancois, L. Endogenous naive CD8+ T cell precursor frequency regulates primary and memory responses to infection. Immunity 2008, 28, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Hand, T.W.; Kaech, S.M. Intrinsic and extrinsic control of effector T cell survival and memory T cell development. Immunol. Res. 2009, 45, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Schluns, K.S.; Kieper, W.C.; Jameson, S.C.; Lefrancois, L. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat. Immunol. 2000, 1, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Tan, J.T.; Wherry, E.J.; Konieczny, B.T.; Surh, C.D.; Ahmed, R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol. 2003, 4, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Huster, K.M.; Busch, V.; Schiemann, M.; Linkemann, K.; Kerksiek, K.M.; Wagner, H.; Busch, D.H. Selective expression of IL-7 receptor on memory T cells identifies early CD40L-dependent generation of distinct CD8+ memory T cell subsets. Proc. Natl. Acad. Sci. USA 2004, 101, 5610–5615. [Google Scholar] [CrossRef] [PubMed]
- Joshi, N.S.; Cui, W.; Chandele, A.; Lee, H.K.; Urso, D.R.; Hagman, J.; Gapin, L.; Kaech, S.M. Inflammation directs memory precursor and short-lived effector CD8+ T cell fates via the graded expression of T-bet transcription factor. Immunity 2007, 27, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Kalia, V.; Haining, W.N.; Konieczny, B.T.; Subramaniam, S.; Ahmed, R. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J. Exp. Med. 2008, 205, 625–640. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, B.S.; Lefrancois, L. Regional and mucosal memory T cells. Nat. Immunol. 2011, 12, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Lefrancois, L.; Obar, J.J. Once a killer, always a killer: From cytotoxic T cell to memory cell. Immunol. Rev. 2010, 235, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Herndler-Brandstetter, D.; Ishigame, H.; Shinnakasu, R.; Plajer, V.; Stecher, C.; Zhao, J.; Lietzenmayer, M.; Kroehling, L.; Takumi, A.; Kometani, K.; et al. Klrg1+ effector CD8+ T cells lose klrg1, differentiate into all memory T cell lineages, and convey enhanced protective immunity. Immunity 2018, 48, 716–729. [Google Scholar] [CrossRef] [PubMed]
- Mercado, R.; Vijh, S.; Allen, S.E.; Kerksiek, K.; Pilip, I.M.; Pamer, E.G. Early programming of T cell populations responding to bacterial infection. J. Immunol. 2000, 165, 6833–6839. [Google Scholar] [CrossRef] [PubMed]
- Badovinac, V.P.; Porter, B.B.; Harty, J.T. Programmed contraction of CD8+ T cells after infection. Nat. Immunol. 2002, 3, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Badovinac, V.P.; Porter, B.B.; Harty, J.T. Cd8+ T cell contraction is controlled by early inflammation. Nat. Immunol. 2004, 5, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Porter, B.B.; Harty, J.T. The onset of CD8+-T-cell contraction is influenced by the peak of Listeria monocytogenes infection and antigen display. Infect. Immun. 2006, 74, 1528–1536. [Google Scholar] [CrossRef] [PubMed]
- Badovinac, V.P.; Harty, J.T. Manipulating the rate of memory CD8+ T cell generation after acute infection. J. Immunol. 2007, 179, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Plumlee, C.R.; Obar, J.J.; Colpitts, S.L.; Jellison, E.R.; Haining, W.N.; Lefrancois, L.; Khanna, K.M. Early effector CD8 T cells display plasticity in populating the short-lived effector and memory-precursor pools following bacterial or viral infection. Sci. Rep. 2015, 5, 12264. [Google Scholar] [CrossRef] [PubMed]
- Plumlee, C.R.; Sheridan, B.S.; Cicek, B.B.; Lefrancois, L. Environmental cues dictate the fate of individual CD8+ T cells responding to infection. Immunity 2013, 39, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Joshi, N.S.; Jiang, A.; Kaech, S.M. Effects of signal 3 during CD8 T cell priming: Bystander production of IL-12 enhances effector T cell expansion but promotes terminal differentiation. Vaccine 2009, 27, 2177–2187. [Google Scholar] [CrossRef] [PubMed]
- Keppler, S.J.; Theil, K.; Vucikuja, S.; Aichele, P. Effector T-cell differentiation during viral and bacterial infections: Role of direct IL-12 signals for cell fate decision of CD8+ T cells. Eur. J. Immunol. 2009, 39, 1774–1783. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, N.; Intlekofer, A.M.; Northrup, J.T.; Wherry, E.J.; Reiner, S.L. Cutting edge: IL-12 inversely regulates T-bet and eomesodermin expression during pathogen-induced CD8+ T cell differentiation. J. Immunol. 2006, 177, 7515–7519. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, M.P.; Lind, N.A.; Purton, J.F.; Filippou, P.; Best, J.A.; McGhee, P.A.; Surh, C.D.; Goldrath, A.W. IL-7 and IL-15 differentially regulate CD8+ T-cell subsets during contraction of the immune response. Blood 2008, 112, 3704–3712. [Google Scholar] [CrossRef] [PubMed]
- Yajima, T.; Yoshihara, K.; Nakazato, K.; Kumabe, S.; Koyasu, S.; Sad, S.; Shen, H.; Kuwano, H.; Yoshikai, Y. IL-15 regulates CD8+ T cell contraction during primary infection. J. Immunol. 2006, 176, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Sanjabi, S.; Mosaheb, M.M.; Flavell, R.A. Opposing effects of TGF-β and IL-15 cytokines control the number of short-lived effector CD8+ T cells. Immunity 2009, 31, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Hand, T.W.; Morre, M.; Kaech, S.M. Expression of IL-7 receptor α is necessary but not sufficient for the formation of memory CD8 T cells during viral infection. Proc. Natl. Acad. Sci. USA 2007, 104, 11730–11735. [Google Scholar] [CrossRef] [PubMed]
- Haring, J.S.; Jing, X.; Bollenbacher-Reilley, J.; Xue, H.H.; Leonard, W.J.; Harty, J.T. Constitutive expression of IL-7 receptor α does not support increased expansion or prevent contraction of antigen-specific CD4 or CD8 T cells following Listeria monocytogenes infection. J. Immunol. 2008, 180, 2855–2862. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Lenig, D.; Forster, R.; Lipp, M.; Lanzavecchia, A. Two subsets of memory t lymphocytes with distinct homing potentials and effector functions. Nature 1999, 401, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Obar, J.J.; Lefrancois, L. Early signals during CD8 T cell priming regulate the generation of central memory cells. J. Immunol. 2010, 185, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Teichgraber, V.; Becker, T.C.; Masopust, D.; Kaech, S.M.; Antia, R.; von Andrian, U.H.; Ahmed, R. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat. Immunol. 2003, 4, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Marzo, A.L.; Klonowski, K.D.; Le Bon, A.; Borrow, P.; Tough, D.F.; Lefrancois, L. Initial T cell frequency dictates memory CD8+ T cell lineage commitment. Nat. Immunol. 2005, 6, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Badovinac, V.P.; Haring, J.S.; Harty, J.T. Initial T cell receptor transgenic cell precursor frequency dictates critical aspects of the CD8+ T cell response to infection. Immunity 2007, 26, 827–841. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.A.; Bevan, M.J. Shortening the infectious period does not alter expansion of CD8 T cells but diminishes their capacity to differentiate into memory cells. J. Immunol. 2004, 173, 6694–6702. [Google Scholar] [CrossRef] [PubMed]
- Klonowski, K.D.; Marzo, A.L.; Williams, K.J.; Lee, S.J.; Pham, Q.M.; Lefrancois, L. Cd8 T cell recall responses are regulated by the tissue tropism of the memory cell and pathogen. J. Immunol. 2006, 177, 6738–6746. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.F.; Wolint, P.; Schwarz, K.; Jager, P.; Oxenius, A. Functional properties and lineage relationship of CD8+ T cell subsets identified by expression of IL-7 receptor α and CD62L. J. Immunol. 2005, 175, 4686–4696. [Google Scholar] [CrossRef] [PubMed]
- Huster, K.M.; Koffler, M.; Stemberger, C.; Schiemann, M.; Wagner, H.; Busch, D.H. Unidirectional development of CD8+ central memory T cells into protective listeria-specific effector memory T cells. Eur. J. Immunol. 2006, 36, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.A.; McDonald-Hyman, C.; Jameson, S.C.; Hamilton, S.E. Effector-like CD8+ T cells in the memory population mediate potent protective immunity. Immunity 2013, 38, 1250–1260. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, T.; Wakim, L.M.; Eidsmo, L.; Reading, P.C.; Heath, W.R.; Carbone, F.R. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat. Immunol. 2009, 10, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Clark, R.A.; Liu, L.; Wagers, A.J.; Fuhlbrigge, R.C.; Kupper, T.S. Skin infection generates non-migratory memory CD8+ TRM cells providing global skin immunity. Nature 2012, 483, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Masopust, D.; Choo, D.; Vezys, V.; Wherry, E.J.; Duraiswamy, J.; Akondy, R.; Wang, J.; Casey, K.A.; Barber, D.L.; Kawamura, K.S.; et al. Dynamic T cell migration program provides resident memory within intestinal epithelium. J. Exp. Med. 2010, 207, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, Y.; Lee, Y.T.; Bouchard, K.R.; Benechet, A.; Khanna, K.; Cauley, L.S. Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J. Leukoc. Biol. 2014, 95, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.K.; Rahimpour, A.; Ma, J.Z.; Collins, N.; Stock, A.T.; Hafon, M.L.; Vega-Ramos, J.; Lauzurica, P.; Mueller, S.N.; Stefanovic, T.; et al. The developmental pathway for CD103+CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 2013, 14, 1294–1301. [Google Scholar] [CrossRef] [PubMed]
- Wakim, L.M.; Woodward-Davis, A.; Liu, R.; Hu, Y.; Villadangos, J.; Smyth, G.; Bevan, M.J. The molecular signature of tissue resident memory CD8 T cells isolated from the brain. J. Immunol. 2012, 189, 3462–3471. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.K.; Minnich, M.; Kragten, N.A.; Liao, Y.; Nota, B.; Seillet, C.; Zaid, A.; Man, K.; Preston, S.; Freestone, D.; et al. Hobit and blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 2016, 352, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Tian, T.; Park, C.O.; Lofftus, S.Y.; Mei, S.; Liu, X.; Luo, C.; O’Malley, J.T.; Gehad, A.; Teague, J.E.; et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 2017, 543, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Mackay, L.K.; Braun, A.; Macleod, B.L.; Collins, N.; Tebartz, C.; Bedoui, S.; Carbone, F.R.; Gebhardt, T. Cutting edge: Cd69 interference with sphingosine-1-phosphate receptor function regulates peripheral T cell retention. J. Immunol. 2015, 194, 2059–2063. [Google Scholar] [CrossRef] [PubMed]
- Skon, C.N.; Lee, J.Y.; Anderson, K.G.; Masopust, D.; Hogquist, K.A.; Jameson, S.C. Transcriptional downregulation of S1PR1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol. 2013, 14, 1285–1293. [Google Scholar] [CrossRef] [PubMed]
- Shiow, L.R.; Rosen, D.B.; Brdickova, N.; Xu, Y.; An, J.; Lanier, L.L.; Cyster, J.G.; Matloubian, M. CD69 acts downstream of interferon-α/β to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 2006, 440, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Bankovich, A.J.; Shiow, L.R.; Cyster, J.G. CD69 suppresses sphingosine 1-Phosophate Receptor-1 (S1P1) function through interaction with membrane helix 4. J. Biol. Chem. 2010, 285, 22328–22337. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.T.; Suarez-Ramirez, J.E.; Wu, T.; Redman, J.M.; Bouchard, K.; Hadley, G.A.; Cauley, L.S. Environmental and antigen receptor-derived signals support sustained surveillance of the lungs by pathogen-specific cytotoxic T lymphocytes. J. Virol. 2011, 85, 4085–4094. [Google Scholar] [CrossRef] [PubMed]
- Casey, K.A.; Fraser, K.A.; Schenkel, J.M.; Moran, A.; Abt, M.C.; Beura, L.K.; Lucas, P.J.; Artis, D.; Wherry, E.J.; Hogquist, K.; et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J. Immunol. 2012, 188, 4866–4875. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A.P.; Clarke, J.; Wood, O.; Garrido-Martin, E.M.; Chee, S.J.; Mellows, T.; Samaniego-Castruita, D.; Singh, D.; Seumois, G.; Alzetani, A.; et al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat. Immunol. 2017, 18, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Malik, B.T.; Byrne, K.T.; Vella, J.L.; Zhang, P.; Shabaneh, T.B.; Steinberg, S.M.; Molodtsov, A.K.; Bowers, J.S.; Angeles, C.V.; Paulos, C.M.; et al. Resident memory T cells in the skin mediate durable immunity to melanoma. Sci. Immunol. 2017, 2, eaam6346. [Google Scholar] [CrossRef] [PubMed]
- Nizard, M.; Roussel, H.; Diniz, M.O.; Karaki, S.; Tran, T.; Voron, T.; Dransart, E.; Sandoval, F.; Riquet, M.; Rance, B.; et al. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat. Commun. 2017, 8, 15221. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, J.M.; Fraser, K.A.; Vezys, V.; Masopust, D. Sensing and alarm function of resident memory CD8+ T cells. Nat. Immunol. 2013, 14, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, J.M.; Fraser, K.A.; Beura, L.K.; Pauken, K.E.; Vezys, V.; Masopust, D. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science 2014, 346, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Ariotti, S.; Hogenbirk, M.A.; Dijkgraaf, F.E.; Visser, L.L.; Hoekstra, M.E.; Song, J.Y.; Jacobs, H.; Haanen, J.B.; Schumacher, T.N. T cell memory. Skin-resident memory CD8+ T cells trigger a state of tissue-wide pathogen alert. Science 2014, 346, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Masopust, D.; Vezys, V.; Marzo, A.L.; Lefrancois, L. Preferential localization of effector memory cells in nonlymphoid tissue. Science 2001, 291, 2413–2417. [Google Scholar] [CrossRef] [PubMed]
- Lefrancois, L.; Parker, C.M.; Olson, S.; Muller, W.; Wagner, N.; Schon, M.P.; Puddington, L. The role of beta7 integrins in CD8 T cell trafficking during an antiviral immune response. J. Exp. Med. 1999, 189, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Johansson-Lindbom, B.; Svensson, M.; Wurbel, M.A.; Malissen, B.; Marquez, G.; Agace, W. Selective generation of gut tropic T cells in gut-associated lymphoid tissue (galt): Requirement for galt dendritic cells and adjuvant. J. Exp. Med. 2003, 198, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Wiesel, M.; Oxenius, A. From crucial to negligible: Functional CD8+ T-cell responses and their dependence on CD4+ T-cell help. Eur. J. Immunol. 2012, 42, 1080–1088. [Google Scholar] [CrossRef] [PubMed]
- Marzo, A.L.; Vezys, V.; Klonowski, K.D.; Lee, S.J.; Muralimohan, G.; Moore, M.; Tough, D.F.; Lefrancois, L. Fully functional memory CD8 T cells in the absence of CD4 T cells. J. Immunol. 2004, 173, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.C.; Bevan, M.J. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science 2003, 300, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.C.; Williams, M.A.; Bevan, M.J. Cd4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection. Nat. Immunol. 2004, 5, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Shedlock, D.J.; Shen, H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science 2003, 300, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Shedlock, D.J.; Whitmire, J.K.; Tan, J.; MacDonald, A.S.; Ahmed, R.; Shen, H. Role of CD4 T cell help and costimulation in CD8 T cell responses during Listeria monocytogenes infection. J. Immunol. 2003, 170, 2053–2063. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, Y.; Lu, B.; Gerard, C.; Iwasaki, A. Cd8+ T lymphocyte mobilization to virus-infected tissue requires CD4+ T-cell help. Nature 2009, 462, 510–513. [Google Scholar] [CrossRef] [PubMed]
- Laidlaw, B.J.; Zhang, N.; Marshall, H.D.; Staron, M.M.; Guan, T.; Hu, Y.; Cauley, L.S.; Craft, J.; Kaech, S.M. Cd4+ T cell help guides formation of CD103+ lung-resident memory CD8+ T cells during influenza viral infection. Immunity 2014, 41, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.C.; Bevan, M.J. Cutting edge: Long-lived CD8 memory and protective immunity in the absence of CD40 expression on CD8 T cells. J. Immunol. 2004, 172, 3385–3389. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.E.; Tvinnereim, A.R.; Harty, J.T. Listeria monocytogenes infection overcomes the requirement for CD40 ligand in exogenous antigen presentation to CD8+ T cells. J. Immunol. 2001, 167, 5603–5609. [Google Scholar] [CrossRef] [PubMed]
- Obar, J.J.; Molloy, M.J.; Jellison, E.R.; Stoklasek, T.A.; Zhang, W.; Usherwood, E.J.; Lefrancois, L. CD4+ T cell regulation of CD25 expression controls development of short-lived effector CD8+ T cells in primary and secondary responses. Proc. Natl. Acad. Sci. USA 2010, 107, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Zenewicz, L.A.; Shen, H. Innate and adaptive immune responses to Listeria monocytogenes: A short overview. Microbes Infect. 2007, 9, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Pamer, E.G.; Harty, J.T.; Bevan, M.J. Precise prediction of a dominant class i mhc-restricted epitope of Listeria monocytogenes. Nature 1991, 353, 852–855. [Google Scholar] [CrossRef] [PubMed]
- Pamer, E.G. Direct sequence identification and kinetic analysis of an MHC class I-restricted Listeria monocytogenes CTL epitope. J. Immunol. 1994, 152, 686–694. [Google Scholar] [PubMed]
- Harty, J.T.; Bevan, M.J. CD8+ T cells specific for a single nonamer epitope of Listeria monocytogenes are protective in vivo. J. Exp. Med. 1992, 175, 1531–1538. [Google Scholar] [CrossRef] [PubMed]
- Harty, J.T.; Pamer, E.G. CD8 T lymphocytes specific for the secreted p60 antigen protect against Listeria monocytogenes infection. J. Immunol. 1995, 154, 4642–4650. [Google Scholar] [PubMed]
- Harty, J.T.; Lenz, L.L.; Bevan, M.J. Primary and secondary immune responses to Listeria monocytogenes. Curr. Opin. Immunol. 1996, 8, 526–530. [Google Scholar] [CrossRef]
- Hess, J.; Gentschev, I.; Miko, D.; Welzel, M.; Ladel, C.; Goebel, W.; Kaufmann, S.H. Superior efficacy of secreted over somatic antigen display in recombinant salmonella vaccine induced protection against listeriosis. Proc. Natl. Acad. Sci. USA 1996, 93, 1458–1463. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Miller, J.F.; Fan, X.; Kolwyck, D.; Ahmed, R.; Harty, J.T. Compartmentalization of bacterial antigens: Differential effects on priming of CD8 T cells and protective immunity. Cell 1998, 92, 535–545. [Google Scholar] [CrossRef]
- Zenewicz, L.A.; Foulds, K.E.; Jiang, J.; Fan, X.; Shen, H. Nonsecreted bacterial proteins induce recall CD8 T cell responses but do not serve as protective antigens. J. Immunol. 2002, 169, 5805–5812. [Google Scholar] [CrossRef] [PubMed]
- Mora-Solano, C.; Wen, Y.; Han, H.; Chen, J.; Chong, A.S.; Miller, M.L.; Pompano, R.R.; Collier, J.H. Active immunotherapy for tnf-mediated inflammation using self-assembled peptide nanofibers. Biomaterials 2017, 149, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Calderon-Gonzalez, R.; Frande-Cabanes, E.; Teran-Navarro, H.; Marimon, J.M.; Freire, J.; Salcines-Cuevas, D.; Carmen Farinas, M.; Onzalez-Rico, C.; Marradi, M.; Garcia, I.; et al. Gnp-gapdh1-22 nanovaccines prevent neonatal listeriosis by blocking microglial apoptosis and bacterial dissemination. Oncotarget 2017, 8, 53916–53934. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Ju, X.; Du, L.; Yuan, J.; Wang, L.; He, R.; Chen, Z. Production of bacterial ghosts from gram-positive pathogen Listeria monocytogenes. Foodborne Pathog. Dis. 2017, 14, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Torres, D.; Kohler, A.; Delbauve, S.; Caminschi, I.; Lahoud, M.H.; Shortman, K.; Flamand, V. IL-12p40/IL-10 producing preCD8α/Clec9A+ dendritic cells are induced in neonates upon Listeria monocytogenes infection. PLoS Pathog. 2016, 12, e1005561. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Zia, Q.; Kazmi, S.; Ahmad, E.; Azhar, A.; Johnson, K.E.; Zubair, S.; Owais, M. Efficacy of cell wall-deficient spheroplasts against experimental murine listeriosis. Scand. J. Immunol. 2015, 82, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Del Rio, E.; Marradi, M.; Calderon-Gonzalez, R.; Frande-Cabanes, E.; Penades, S.; Petrovsky, N.; Alvarez-Dominguez, C. A gold glyco-nanoparticle carrying a listeriolysin O peptide and formulated with advax delta inulin adjuvant induces robust T-cell protection against listeria infection. Vaccine 2015, 33, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.R.; Chaturvedi, V.; Kinder, J.M.; Jiang, T.T.; Xin, L.; Ertelt, J.M.; Way, S.S. Perinatal Listeria monocytogenes susceptibility despite preconceptual priming and maintenance of pathogen-specific CD8+ T cells during pregnancy. Cell. Mol. Immunol. 2014, 11, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Nakamura, Y.; Suda, T.; Uchijima, M.; Tsujimura, K.; Nagata, T.; Giermasz, A.S.; Kalinski, P.; Nakamura, H.; Chida, K. Enhancement of protective immunity against intracellular bacteria using type-1 polarized dendritic cell (DC) vaccine. Vaccine 2012, 30, 2633–2639. [Google Scholar] [CrossRef] [PubMed]
- Masopust, D.; Vezys, V.; Wherry, E.J.; Barber, D.L.; Ahmed, R. Cutting edge: Gut microenvironment promotes differentiation of a unique memory CD8 T cell population. J. Immunol. 2006, 176, 2079–2083. [Google Scholar] [CrossRef] [PubMed]
- Masopust, D.; Ha, S.J.; Vezys, V.; Ahmed, R. Stimulation history dictates memory CD8 T cell phenotype: Implications for prime-boost vaccination. J. Immunol. 2006, 177, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Schenkel, J.M.; Fraser, K.A.; Casey, K.A.; Beura, L.K.; Pauken, K.E.; Vezys, V.; Masopust, D. IL-15-independent maintenance of tissue-resident and boosted effector memory CD8 T cells. J. Immunol. 2016, 196, 3920–3926. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Iwasaki, A. A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature 2012, 491, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Lecuit, M.; Nelson, D.M.; Smith, S.D.; Khun, H.; Huerre, M.; Vacher-Lavenu, M.C.; Gordon, J.I.; Cossart, P. Targeting and crossing of the human maternofetal barrier by Listeria monocytogenes: Role of internalin interaction with trophoblast e-cadherin. Proc. Natl. Acad. Sci. USA 2004, 101, 6152–6157. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, V.; Ertelt, J.M.; Jiang, T.T.; Kinder, J.M.; Xin, L.; Owens, K.J.; Jones, H.N.; Way, S.S. CXCR3 blockade protects against Listeria monocytogenes infection-induced fetal wastage. J. Clin. Investig. 2015, 125, 1713–1725. [Google Scholar] [CrossRef] [PubMed]
- Pamer, E.G.; Wang, C.R.; Flaherty, L.; Lindahl, K.F.; Bevan, M.J. H-2M3 presents a Listeria monocytogenes peptide to cytotoxic T lymphocytes. Cell 1992, 70, 215–223. [Google Scholar] [CrossRef]
- Kurlander, R.J.; Shawar, S.M.; Brown, M.L.; Rich, R.R. Specialized role for a murine class i-b MHC molecule in prokaryotic host defenses. Science 1992, 257, 678–679. [Google Scholar] [CrossRef] [PubMed]
- Lenz, L.L.; Dere, B.; Bevan, M.J. Identification of an H2-M3-restricted listeria epitope: Implications for antigen presentation by M3. Immunity 1996, 5, 63–72. [Google Scholar] [CrossRef]
- Princiotta, M.F.; Lenz, L.L.; Bevan, M.J.; Staerz, U.D. H2-M3 restricted presentation of a listeria-derived leader peptide. J. Exp. Med. 1998, 187, 1711–1719. [Google Scholar] [CrossRef] [PubMed]
- Gulden, P.H.; Fischer, P., 3rd; Sherman, N.E.; Wang, W.; Engelhard, V.H.; Shabanowitz, J.; Hunt, D.F.; Pamer, E.G. A Listeria monocytogenes pentapeptide is presented to cytolytic T lymphocytes by the H2-M3 mhc class ib molecule. Immunity 1996, 5, 73–79. [Google Scholar] [CrossRef]
- Kerksiek, K.M.; Busch, D.H.; Pamer, E.G. Variable immunodominance hierarchies for H2-M3-restricted n-formyl peptides following bacterial infection. J. Immunol. 2001, 166, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Ploss, A.; Lauvau, G.; Contos, B.; Kerksiek, K.M.; Guirnalda, P.D.; Leiner, I.; Lenz, L.L.; Bevan, M.J.; Pamer, E.G. Promiscuity of mhc class ib-restricted T cell responses. J. Immunol. 2003, 171, 5948–5955. [Google Scholar] [CrossRef] [PubMed]
- D’Orazio, S.E.; Velasquez, M.; Roan, N.R.; Naveiras-Torres, O.; Starnbach, M.N. The Listeria monocytogenes lema gene product is not required for intracellular infection or to activate fmigwii-specific T cells. Infect. Immun. 2003, 71, 6721–6727. [Google Scholar] [CrossRef] [PubMed]
- Ploss, A.; Tran, A.; Menet, E.; Leiner, I.; Pamer, E.G. Cross-recognition of n-formylmethionine peptides is a general characteristic of H2-M3-restricted CD8+ T cells. Infect. Immun. 2005, 73, 4423–4426. [Google Scholar] [CrossRef] [PubMed]
- Seaman, M.S.; Wang, C.R.; Forman, J. MHC class ib-restricted CTL provide protection against primary and secondary Listeria monocytogenes infection. J. Immunol. 2000, 165, 5192–5201. [Google Scholar] [CrossRef] [PubMed]
- Kerksiek, K.M.; Busch, D.H.; Pilip, I.M.; Allen, S.E.; Pamer, E.G. H2-M3-restricted T cells in bacterial infection: Rapid primary but diminished memory responses. J. Exp. Med. 1999, 190, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Chun, T.; Choi, H.J.; Wang, B.; Wang, C.R. Impaired response to listeria in H2-M3-deficient mice reveals a nonredundant role of MHC class ib-specific T cells in host defense. J. Exp. Med. 2006, 203, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Kerksiek, K.M.; Ploss, A.; Leiner, I.; Busch, D.H.; Pamer, E.G. H2-m3-restricted memory T cells: Persistence and activation without expansion. J. Immunol. 2003, 170, 1862–1869. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.E.; Porter, B.B.; Messingham, K.A.; Badovinac, V.P.; Harty, J.T. Mhc class IA-restricted memory T cells inhibit expansion of a nonprotective mhc class ib (H2-M3)-restricted memory response. Nat. Immunol. 2004, 5, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Rothman, J.; Paterson, Y. Live-attenuated listeria-based immunotherapy. Expert Rev. Vaccines 2013, 12, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.M.; Paterson, Y. Attenuated Listeria monocytogenes: A powerful and versatile vector for the future of tumor immunotherapy. Front. Cell. Infect. Microbiol. 2014, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.A.; Shabahang, S.; Timiryasova, T.M.; Zhang, Q.; Beltz, R.; Gentschev, I.; Goebel, W.; Szalay, A.A. Visualization of tumors and metastases in live animals with bacteria and vaccinia virus encoding light-emitting proteins. Nat. Biotechnol. 2004, 22, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Castro, F.; Paterson, Y.; Gravekamp, C. High efficacy of a listeria-based vaccine against metastatic breast cancer reveals a dual mode of action. Cancer Res. 2009, 69, 5860–5866. [Google Scholar] [CrossRef] [PubMed]
- Quispe-Tintaya, W.; Chandra, D.; Jahangir, A.; Harris, M.; Casadevall, A.; Dadachova, E.; Gravekamp, C. Nontoxic radioactive listeriaat is a highly effective therapy against metastatic pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 8668–8673. [Google Scholar] [CrossRef] [PubMed]
- Finelli, A.; Kerksiek, K.M.; Allen, S.E.; Marshall, N.; Mercado, R.; Pilip, I.; Busch, D.H.; Pamer, E.G. Mhc class I restricted T cell responses to Listeria monocytogenes, an intracellular bacterial pathogen. Immunol. Res. 1999, 19, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Wolf, B.J.; Princiotta, M.F. Processing of recombinant Listeria monocytogenes proteins for mhc class I presentation follows a dedicated, high-efficiency pathway. J. Immunol. 2013, 190, 2501–2509. [Google Scholar] [CrossRef] [PubMed]
- Souders, N.C.; Sewell, D.A.; Pan, Z.K.; Hussain, S.F.; Rodriguez, A.; Wallecha, A.; Paterson, Y. Listeria-based vaccines can overcome tolerance by expanding low avidity CD8+ T cells capable of eradicating a solid tumor in a transgenic mouse model of cancer. Cancer Immun. 2007, 7, 2. [Google Scholar] [PubMed]
- Sewell, D.A.; Pan, Z.K.; Paterson, Y. Listeria-based HPV-16 E7 vaccines limit autochthonous tumor growth in a transgenic mouse model for HPV-16 transformed tumors. Vaccine 2008, 26, 5315–5320. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.F.; Paterson, Y. CD4+CD25+ regulatory T cells that secrete tgfbeta and IL-10 are preferentially induced by a vaccine vector. J. Immunother. 2004, 27, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Wallecha, A.; Singh, R.; Malinina, I. Listeria monocytogenes (Lm)-LLO immunotherapies reduce the immunosuppressive activity of myeloid-derived suppressor cells and regulatory T cells in the tumor microenvironment. J. Immunother. 2013, 36, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Mkrtichyan, M.; Chong, N.; Abu Eid, R.; Wallecha, A.; Singh, R.; Rothman, J.; Khleif, S.N. Anti-pd-1 antibody significantly increases therapeutic efficacy of Listeria monocytogenes (Lm)-LLO immunotherapy. J. Immunother. Cancer 2013, 1, 15. [Google Scholar] [CrossRef] [PubMed]
- Guirnalda, P.; Wood, L.; Goenka, R.; Crespo, J.; Paterson, Y. Interferon gamma-induced intratumoral expression of cxcl9 alters the local distribution of T cells following immunotherapy with Listeria monocytogenes. Oncoimmunology 2013, 2, e25752. [Google Scholar] [CrossRef] [PubMed]
- Paterson, Y.; Johnson, R.S. Progress towards the use of Listeria monocytogenes as a live bacterial vaccine vector for the delivery of HIV antigens. Expert Rev. Vaccines 2004, 3, S119–S134. [Google Scholar] [CrossRef] [PubMed]
- Cory, L.; Chu, C. ADXS-HPV: A therapeutic listeria vaccination targeting cervical cancers expressing the HPV E7 antigen. Hum. Vaccines Immunother. 2014, 10, 3190–3195. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Brockstedt, D.G.; Nir-Paz, R.; Hampl, J.; Mathur, S.; Nemunaitis, J.; Sterman, D.H.; Hassan, R.; Lutz, E.; Moyer, B.; et al. A live-attenuated listeria vaccine (ANZ-100) and a live-attenuated listeria vaccine expressing mesothelin (CRS-207) for advanced cancers: Phase i studies of safety and immune induction. Clin. Cancer Res. 2012, 18, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Wang-Gillam, A.; Picozzi, V.; Greten, T.F.; Crocenzi, T.; Springett, G.; Morse, M.; Zeh, H.; Cohen, D.; Fine, R.L.; et al. Safety and survival with gvax pancreas prime and Listeria monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J. Clin. Oncol. 2015, 33, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- Maciag, P.C.; Radulovic, S.; Rothman, J. The first clinical use of a live-attenuated Listeria monocytogenes vaccine: A phase I safety study of LM-LLO-E7 in patients with advanced carcinoma of the cervix. Vaccine 2009, 27, 3975–3983. [Google Scholar] [CrossRef] [PubMed]
- Mason, N.J.; Gnanandarajah, J.S.; Engiles, J.B.; Gray, F.; Laughlin, D.; Gaurnier-Hausser, A.; Wallecha, A.; Huebner, M.; Paterson, Y. Immunotherapy with a her2-targeting listeria induces her2-specific immunity and demonstrates potential therapeutic effects in a phase I trial in canine osteosarcoma. Clin. Cancer Res. 2016, 22, 4380–4390. [Google Scholar] [CrossRef] [PubMed]
- Pages, F.; Galon, J.; Dieu-Nosjean, M.C.; Tartour, E.; Sautes-Fridman, C.; Fridman, W.H. Immune infiltration in human tumors: A prognostic factor that should not be ignored. Oncogene 2010, 29, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Gooden, M.J.; de Bock, G.H.; Leffers, N.; Daemen, T.; Nijman, H.W. The prognostic influence of tumour-infiltrating lymphocytes in cancer: A systematic review with meta-analysis. Br. J. Cancer 2011, 105, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Huang, H.; Grenier, J.M.; Perez, O.A.; Smilowitz, H.M.; Adler, B.; Khanna, K.M. Cytomegalovirus-based vaccine expressing a modified tumor antigen induces potent tumor-specific CD8+ T-cell response and protects mice from melanoma. Cancer Immunol. Res. 2015, 3, 536–546. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, Z.; Khairallah, C.; Sheridan, B.S. Listeria Monocytogenes: A Model Pathogen Continues to Refine Our Knowledge of the CD8 T Cell Response. Pathogens 2018, 7, 55. https://doi.org/10.3390/pathogens7020055
Qiu Z, Khairallah C, Sheridan BS. Listeria Monocytogenes: A Model Pathogen Continues to Refine Our Knowledge of the CD8 T Cell Response. Pathogens. 2018; 7(2):55. https://doi.org/10.3390/pathogens7020055
Chicago/Turabian StyleQiu, Zhijuan, Camille Khairallah, and Brian S. Sheridan. 2018. "Listeria Monocytogenes: A Model Pathogen Continues to Refine Our Knowledge of the CD8 T Cell Response" Pathogens 7, no. 2: 55. https://doi.org/10.3390/pathogens7020055
APA StyleQiu, Z., Khairallah, C., & Sheridan, B. S. (2018). Listeria Monocytogenes: A Model Pathogen Continues to Refine Our Knowledge of the CD8 T Cell Response. Pathogens, 7(2), 55. https://doi.org/10.3390/pathogens7020055