Whole Genome Classification and Phylogenetic Analyses of Rotavirus B strains from the United States


Abstract
1. Introduction
2. Materials and Methods
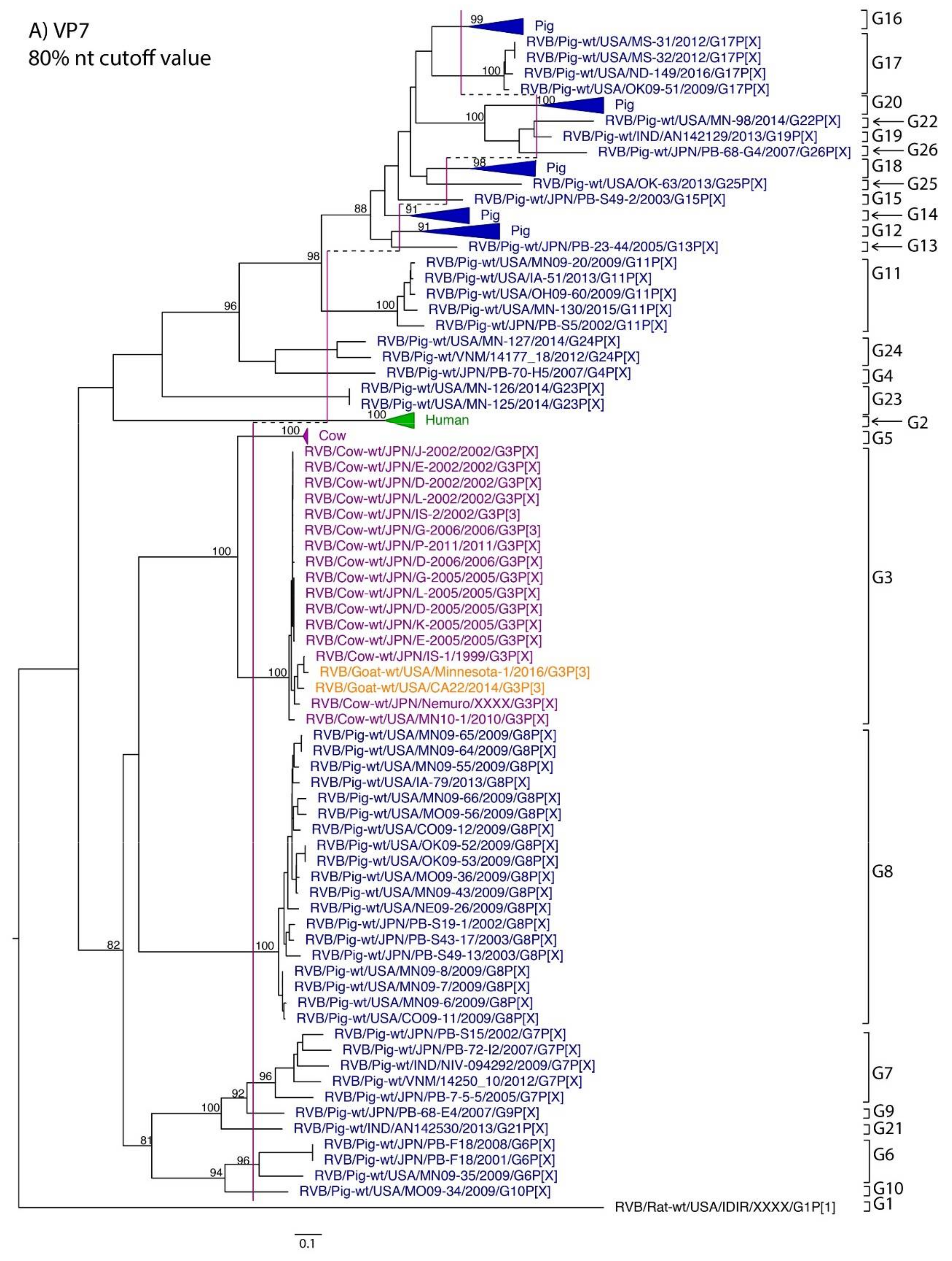
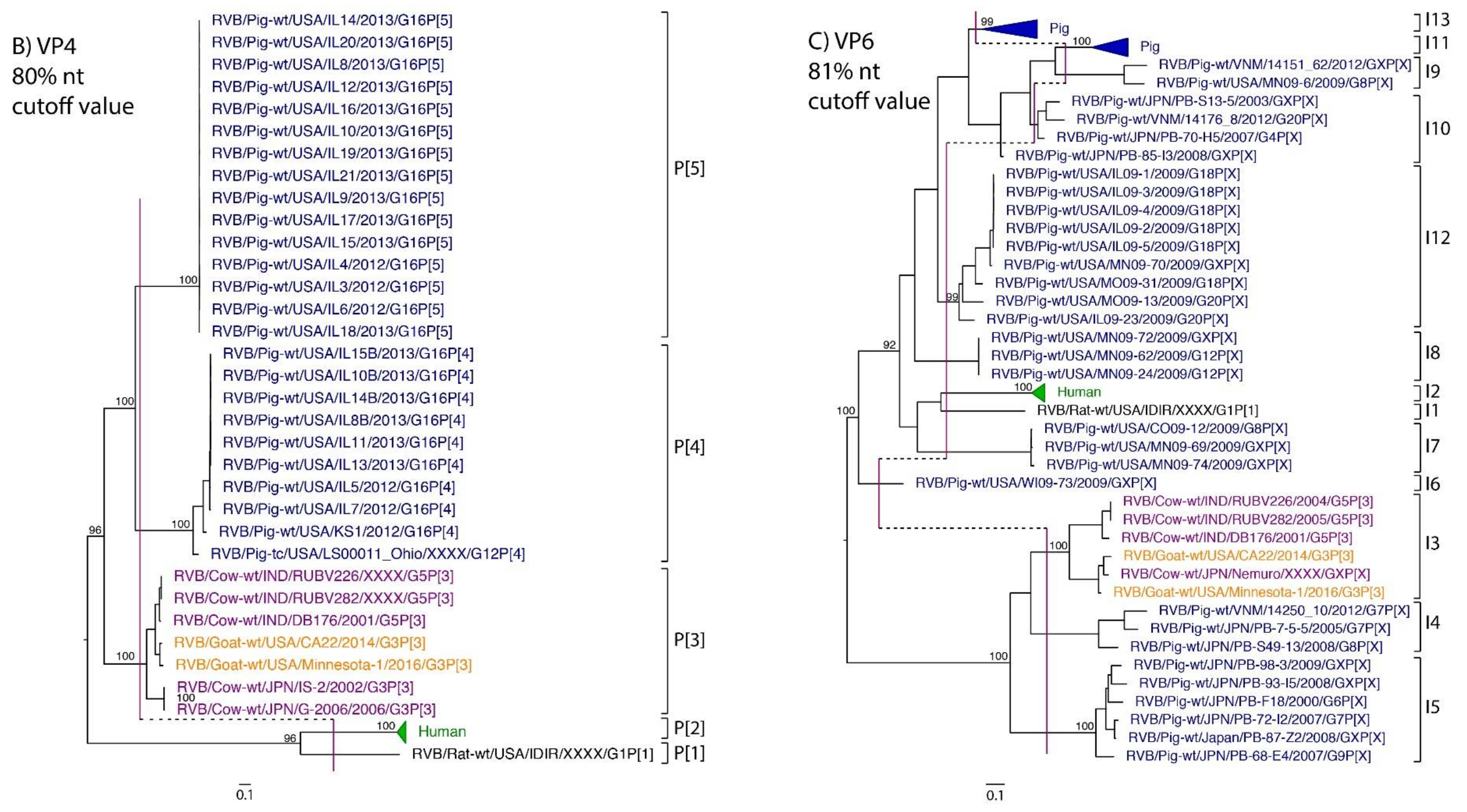
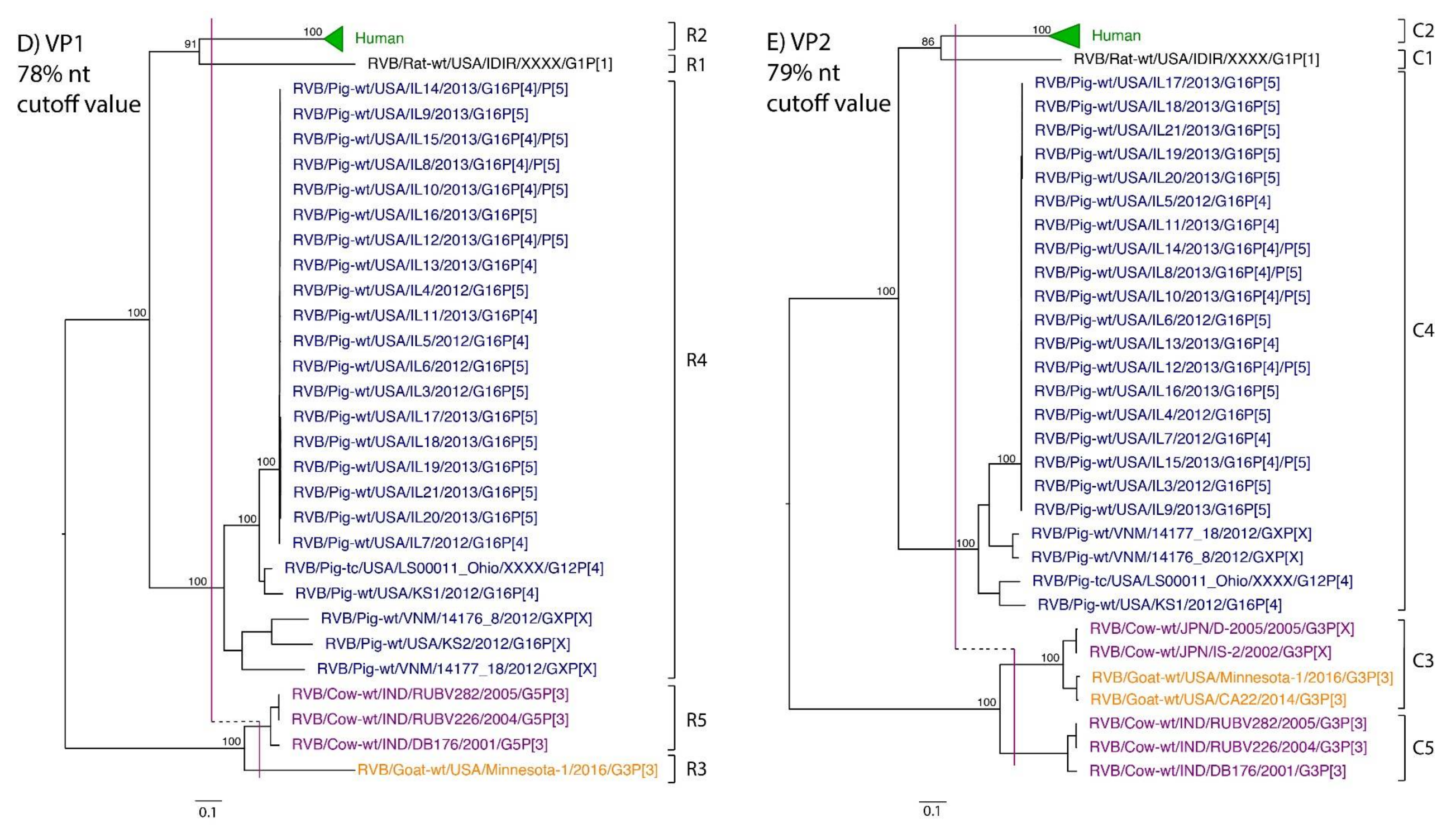
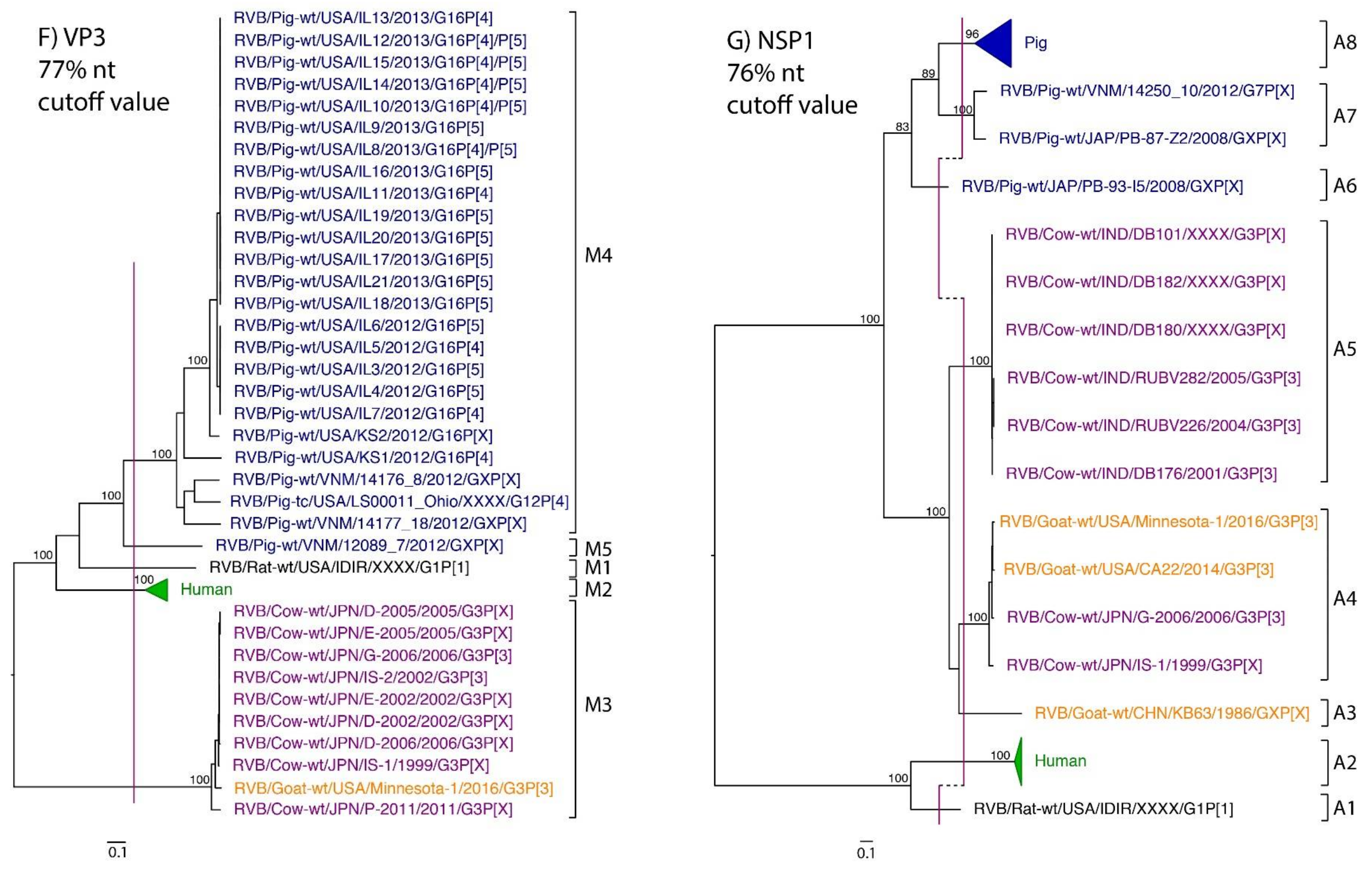
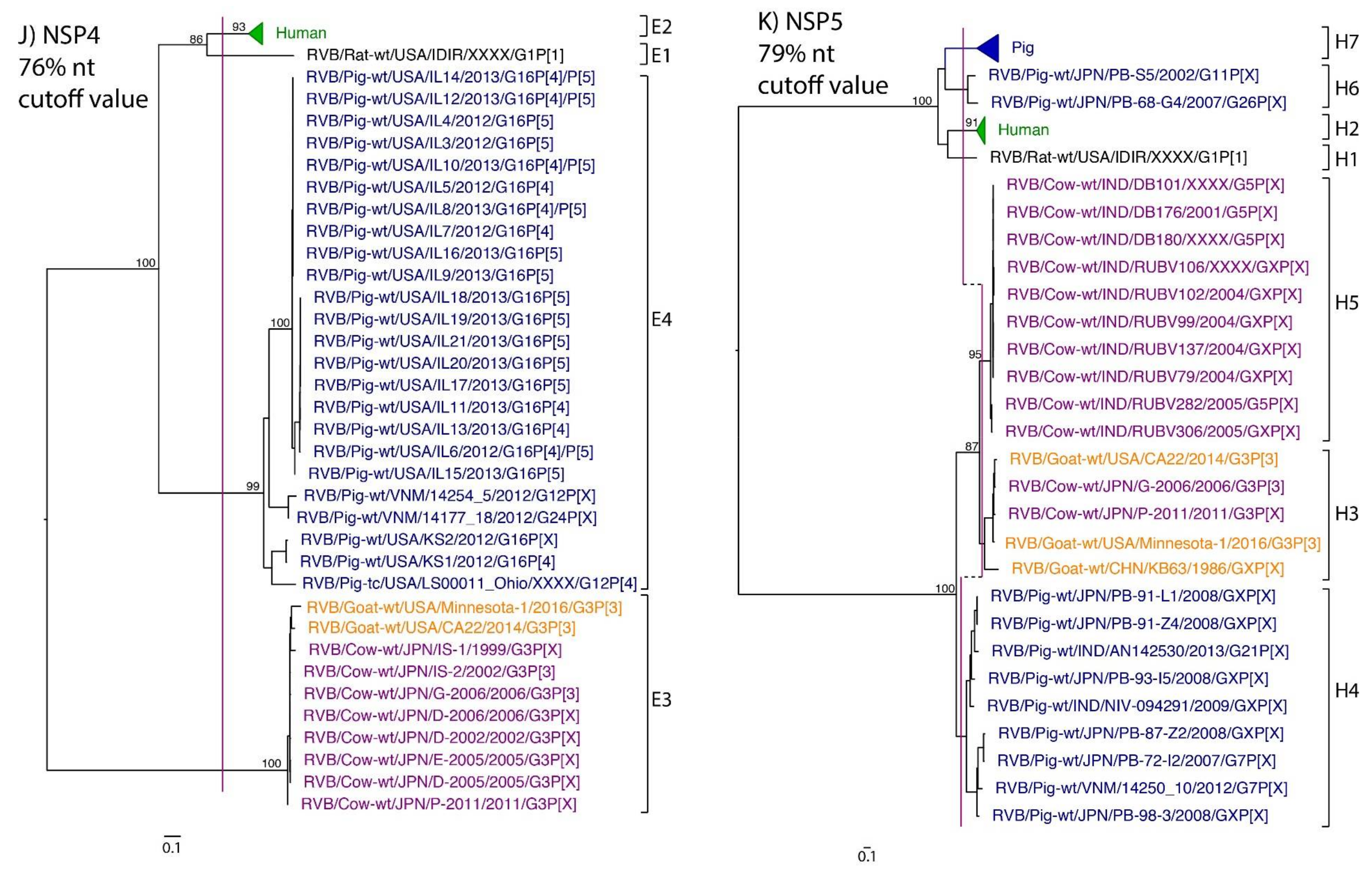
3. Results
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Saif, L.J.; Jiang, B. Nongroup A rotaviruses of humans and animals. Curr. Top. Microbiol. Immunol. 1994, 185, 339–371. [Google Scholar] [PubMed]
- Martella, V.; Bányai, K.; Matthijnssens, J.; Buonavoglia, C.; Ciarlet, M. Zoonotic aspects of rotaviruses. Vet. Microbiol. 2010, 140, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Estes, M.K.; Greenberg, H.B. Rotaviruses, Fields Virology, 6th ed.; Lippincott, Williams & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Matthijnssens, J.; Otto, P.H.; Ciarlet, M.; Desselberger, U.; Van Ranst, M.; Johne, R. VP6-sequence-based cutoff values as a criterion for rotavirus species demarcation. Arch. Virol. 2012, 157, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Mihalov-Kovács, E.; Gellért, Á.; Marton, S.; Farkas, S.L.; Fehér, E.; Oldal, M.; Jakab, F.; Martella, V.; Bányai, K. Candidate new rotavirus species in sheltered dogs, Hungary. Emerg. Infect. Dis. 2015, 21, 660–663. [Google Scholar] [CrossRef] [PubMed]
- Bányai, K.; Kemenesi, G.; Budinski, I.; Földes, F.; Zana, B.; Marton, S.; Varga-Kugler, R.; Oldal, M.; Kurucz, K.; Jakab, F. Candidate new rotavirus species in Schreiber’s bats, Serbia. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2017, 48, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Matthijnssens, J.; Martella, V.; Van Ranst, M. Genomic evolution, host-species barrier, reassortment and classification of rotaviruses. Future Virol. 2010, 5, 385–390. [Google Scholar] [CrossRef]
- Hung, T.; Chen, G.M.; Wang, C.G.; Yao, H.L.; Fang, Z.Y.; Chao, T.X.; Chou, Z.Y.; Ye, W.; Chang, X.J.; Den, S.S. Waterborne outbreak of rotavirus diarrhoea in adults in China caused by a novel rotavirus. Lancet Lond. Engl. 1984, 1, 1139–1142. [Google Scholar]
- Chen, C.M.; Hung, T.; Bridger, J.C.; McCrae, M.A. Chinese adult rotavirus is a group B rotavirus. Lancet Lond. Engl. 1985, 2, 1123–1124. [Google Scholar] [CrossRef]
- Krishnan, T.; Sen, A.; Choudhury, J.S.; Das, S.; Naik, T.N.; Bhattacharya, S.K. Emergence of adult diarrhoea rotavirus in Calcutta, India. Lancet Lond. Engl. 1999, 353, 380–381. [Google Scholar] [CrossRef]
- Kelkar, S.D.; Zade, J.K. Group B rotaviruses similar to strain CAL-1, have been circulating in Western India since 1993. Epidemiol. Infect. 2004, 132, 745–749. [Google Scholar] [CrossRef] [PubMed]
- Sanekata, T.; Ahmed, M.U.; Kader, A.; Taniguchi, K.; Kobayashi, N. Human group B rotavirus infections cause severe diarrhea in children and adults in Bangladesh. J. Clin. Microbiol. 2003, 41, 2187–2190. [Google Scholar] [CrossRef] [PubMed]
- Aung, T.S.; Kobayashi, N.; Nagashima, S.; Ghosh, S.; Aung, M.S.; Oo, K.Y.; Win, N. Detection of group B rotavirus in an adult with acute gastroenteritis in Yangon, Myanmar. J. Med. Virol. 2009, 81, 1968–1974. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.S.; Ganorkar, N.N.; Ranshing, S.S.; Basu, A.; Chavan, N.A.; Gopalkrishna, V. Identification of group B rotavirus as an etiological agent in the gastroenteritis outbreak in Maharashtra, India. J. Med. Virol. 2017, 89, 2244–2248. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.M.; Pun, S.B.; Gauchan, P.; Yokoo, M.; Doan, Y.H.; Tran, T.N.H.; Nakagomi, T.; Nakagomi, O.; Pandey, B.D. The First Identification of Rotavirus B from Children and Adults with Acute Diarrhoea in Kathmandu, Nepal. Trop. Med. Health 2013, 41, 129–134. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Eiden, J.J.; Nataro, J.; Vonderfecht, S.; Petric, M. Molecular cloning, sequence analysis, in vitro expression, and immunoprecipitation of the major inner capsid protein of the IDIR strain of group B rotavirus (GBR). Virology 1992, 188, 580–589. [Google Scholar] [CrossRef]
- Barman, P.; Ghosh, S.; Das, S.; Varghese, V.; Chaudhuri, S.; Sarkar, S.; Krishnan, T.; Bhattacharya, S.K.; Chakrabarti, A.; Kobayashi, N.; et al. Sequencing and sequence analysis of VP7 and NSP5 genes reveal emergence of a new genotype of bovine group B rotaviruses in India. J. Clin. Microbiol. 2004, 42, 2816–2818. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.O.; Parwani, A.V.; Smith, D.; Saif, L.J. Detection of group B rotaviruses in fecal samples from diarrheic calves and adult cows and characterization of their VP7 genes. J. Clin. Microbiol. 1997, 35, 2107–2110. [Google Scholar] [PubMed]
- Chinsangaram, J.; Schore, C.E.; Guterbock, W.; Weaver, L.D.; Osburn, B.I. Prevalence of group A and group B rotaviruses in the feces of neonatal dairy calves from California. Comp. Immunol. Microbiol. Infect. Dis. 1995, 18, 93–103. [Google Scholar] [CrossRef]
- Ghosh, S.; Varghese, V.; Sinha, M.; Kobayashi, N.; Naik, T.N. Evidence for interstate transmission and increase in prevalence of bovine group B rotavirus strains with a novel VP7 genotype among diarrhoeic calves in Eastern and Northern states of India. Epidemiol. Infect. 2007, 135, 1324–1330. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Murakami, T.; Kuroda, Y.; Takai, H.; Ide, H.; Awang, A.; Suzuki, T.; Miyazaki, A.; Nagai, M.; Tsunemitsu, H. Reinfection of adult cattle with rotavirus B during repeated outbreaks of epidemic diarrhea. Can. J. Vet. Res. 2016, 80, 189–196. [Google Scholar] [PubMed]
- Tsunemitsu, H.; Morita, D.; Takaku, H.; Nishimori, T.; Imai, K.; Saif, L.J. First detection of bovine group B rotavirus in Japan and sequence of its VP7 gene. Arch. Virol. 1999, 144, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; McKee, T.A.; Wang, Z.D.; Desselberger, U.; Liu, D.X. Sequence analysis and in vitro expression of genes 6 and 11 of an ovine group B rotavirus isolate, KB63: Evidence for a non-defective, C-terminally truncated NSP1 and a phosphorylated NSP5. J. Gen. Virol. 1999, 80 Pt 8, 2077–2085. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Knutson, T.P.; Ciarlet, M.; Sturos, M.; Marthaler, D.G. Complete genome characterization of a rotavirus B (RVB) strain identified in Alpine goat kids with enteritis reveals inter-species transmission with RVB bovine strains. J. Gen. Virol. 2018, 99, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Theil, K.W.; Grooms, D.L.; McCloskey, C.M.; Redman, D.R. Group B rotavirus associated with an outbreak of neonatal lamb diarrhea. J. Vet. Diagn. Investig. Off. Publ. Am. Assoc. Vet. Lab. Diagn. Inc 1995, 7, 148–150. [Google Scholar] [CrossRef] [PubMed]
- Chasey, D.; Bridger, J.C.; McCrae, M.A. A new type of atypical rotavirus in pigs. Arch. Virol. 1986, 89, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Kuga, K.; Miyazaki, A.; Suzuki, T.; Takagi, M.; Hattori, N.; Katsuda, K.; Mase, M.; Sugiyama, M.; Tsunemitsu, H. Genetic diversity and classification of the outer capsid glycoprotein VP7 of porcine group B rotaviruses. Arch. Virol. 2009, 154, 1785–1795. [Google Scholar] [CrossRef] [PubMed]
- Marthaler, D.; Rossow, K.; Gramer, M.; Collins, J.; Goyal, S.; Tsunemitsu, H.; Kuga, K.; Suzuki, T.; Ciarlet, M.; Matthijnssens, J. Detection of substantial porcine group B rotavirus genetic diversity in the United States, resulting in a modified classification proposal for G genotypes. Virology 2012, 433, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Matthijnssens, J.; Ciarlet, M.; Heiman, E.; Arijs, I.; Delbeke, T.; McDonald, S.M.; Palombo, E.A.; Iturriza-Gómara, M.; Maes, P.; Patton, J.T.; et al. Full genome-based classification of rotaviruses reveals a common origin between human Wa-Like and porcine rotavirus strains and human DS-1-like and bovine rotavirus strains. J. Virol. 2008, 82, 3204–3219. [Google Scholar] [CrossRef] [PubMed]
- Hayashi-Miyamoto, M.; Murakami, T.; Minami-Fukuda, F.; Tsuchiaka, S.; Kishimoto, M.; Sano, K.; Naoi, Y.; Asano, K.; Ichimaru, T.; Haga, K.; et al. Diversity in VP3, NSP3, and NSP4 of rotavirus B detected from Japanese cattle. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2017, 49, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Marthaler, D.; Suzuki, T.; Rossow, K.; Culhane, M.; Collins, J.; Goyal, S.; Tsunemitsu, H.; Ciarlet, M.; Matthijnssens, J. VP6 genetic diversity, reassortment, intragenic recombination and classification of rotavirus B in American and Japanese pigs. Vet. Microbiol. 2014, 172, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Soma, J.; Kuga, K.; Miyazaki, A.; Tsunemitsu, H. Sequence and phylogenetic analyses of nonstructural protein 2 genes of species B porcine rotaviruses detected in Japan during 2001–2009. Virus Res. 2012, 165, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Soma, J.; Miyazaki, A.; Tsunemitsu, H. Phylogenetic analysis of nonstructural protein 5 (NSP5) gene sequences in porcine rotavirus B strains. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2012, 12, 1661–1668. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Kuga, K.; Miyazaki, A.; Tsunemitsu, H. Genetic divergence and classification of non-structural protein 1 among porcine rotaviruses of species B. J. Gen. Virol. 2011, 92, 2922–2929. [Google Scholar] [CrossRef] [PubMed]
- Lahon, A.; Ingle, V.C.; Birade, H.S.; Raut, C.G.; Chitambar, S.D. Molecular characterization of group B rotavirus circulating in pigs from India: Identification of a strain bearing a novel VP7 genotype, G21. Vet. Microbiol. 2014, 174, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Lambden, P.R.; Cooke, S.J.; Caul, E.O.; Clarke, I.N. Cloning of noncultivatable human rotavirus by single primer amplification. J. Virol. 1992, 66, 1817–1822. [Google Scholar] [PubMed]
- Maan, S.; Rao, S.; Maan, N.S.; Anthony, S.J.; Attoui, H.; Samuel, A.R.; Mertens, P.P.C. Rapid cDNA synthesis and sequencing techniques for the genetic study of bluetongue and other dsRNA viruses. J. Virol. Methods 2007, 143, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Hasebe, A. A provisional complete genome-based genotyping system for rotavirus species C from terrestrial mammals. J. Gen. Virol. 2017, 98, 2647–2662. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Navarro, R.; Malik, Y.S.; Willingham, A.L.; Kobayashi, N. Whole genomic analysis of a porcine G6P[13] rotavirus strain. Vet. Microbiol. 2015, 180, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Vlasova, A.N.; Amimo, J.O.; Saif, L.J. Porcine Rotaviruses: Epidemiology, Immune Responses and Control Strategies. Viruses 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.-J.; Matthijnssens, J.; Kim, D.-S.; Kim, J.-Y.; Alfajaro, M.M.; Park, J.-G.; Hosmillo, M.; Son, K.-Y.; Soliman, M.; Baek, Y.-B.; et al. Genetic diversity of the VP7, VP4 and VP6 genes of Korean porcine group C rotaviruses. Vet. Microbiol. 2015, 176, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Marthaler, D.; Rossow, K.; Culhane, M.; Collins, J.; Goyal, S.; Ciarlet, M.; Matthijnssens, J. Identification, phylogenetic analysis and classification of porcine group C rotavirus VP7 sequences from the United States and Canada. Virology 2013, 446, 189–198. [Google Scholar] [CrossRef] [PubMed]
- McDonald, S.M.; Matthijnssens, J.; McAllen, J.K.; Hine, E.; Overton, L.; Wang, S.; Lemey, P.; Zeller, M.; Van Ranst, M.; Spiro, D.J.; et al. Evolutionary Dynamics of Human Rotaviruses: Balancing Reassortment with Preferred Genome Constellations. PLoS Pathog. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Steyer, A.; Poljsak-Prijatelj, M.; Barlic-Maganja, D.; Marin, J. Human, porcine and bovine rotaviruses in Slovenia: Evidence of interspecies transmission and genome reassortment. J. Gen. Virol. 2008, 89, 1690–1698. [Google Scholar] [CrossRef] [PubMed]
- Aung, M.S.; Nahar, S.; Aida, S.; Paul, S.K.; Hossain, M.A.; Ahmed, S.; Haque, N.; Ghosh, S.; Malik, Y.S.; Urushibara, N.; et al. Distribution of two distinct rotavirus B (RVB) strains in the north-central Bangladesh and evidence for reassortment event among human RVB revealed by whole genomic analysis. Infect. Genet. Evol. 2017, 47, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Agbemabiese, C.A.; Nakagomi, T.; Gauchan, P.; Sherchand, J.B.; Pandey, B.D.; Cunliffe, N.A.; Nakagomi, O. Whole genome characterisation of a porcine-like human reassortant G26P[19] Rotavirus A strain detected in a child hospitalised for diarrhoea in Nepal, 2007. Infect. Genet. Evol. 2017, 54, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Pietsch, C.; Liebert, U.G. Molecular characterization of different equine-like G3 rotavirus strains from Germany. Infect. Genet. Evol. 2018, 57, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Bwogi, J.; Jere, K.C.; Karamagi, C.; Byarugaba, D.K.; Namuwulya, P.; Baliraine, F.N.; Desselberger, U.; Iturriza-Gomara, M. Whole genome analysis of selected human and animal rotaviruses identified in Uganda from 2012 to 2014 reveals complex genome reassortment events between human, bovine, caprine and porcine strains. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Alam, M.M.; Ahmed, M.U.; Talukdar, R.I.; Paul, S.K.; Kobayashi, N. Complete genome constellation of a caprine group A rotavirus strain reveals common evolution with ruminant and human rotavirus strains. J. Gen. Virol. 2010, 91, 2367–2373. [Google Scholar] [CrossRef] [PubMed]
- Louge Uriarte, E.L.; Badaracco, A.; Matthijnssens, J.; Zeller, M.; Heylen, E.; Manazza, J.; Miño, S.; Van Ranst, M.; Odeón, A.; Parreño, V. The first caprine rotavirus detected in Argentina displays genomic features resembling virus strains infecting members of the Bovidae and Camelidae. Vet. Microbiol. 2014, 171, 189–197. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Segment | Porcine | Bovine | Human | |||
|---|---|---|---|---|---|---|
| Range | Median | Range | Median | Range | Median | |
| VP7 | 55.0–100 | 74.0 b | 77.0–100 | 94.0 a | 91.0–100 | 98.0 c |
| VP4 | 64.0–100 | 75.5 a | 81.0–100 | 82.0 ab | 90.0–100 | 98.0 b |
| VP6 | 63.0–100 | 77.0 b | 82.0–100 | 87.0 a | 92.0–100 | 98.0 a |
| VP1 | 78.0–100 | 100.0 a | 94.0–100 | 94.0 ab | 90.0–100 | 98.0 b |
| VP2 | 82.0–100 | 100 a | 77.0–100 | 77.0 ab | 90.0–100 | 98.0 b |
| VP3 | 72.0–100 | 97.0 b | 92.0–100 | 99.0 a | 90.0–100 | 98.0 a |
| NSP1 | 67.0–100 | 83.0 a | 71.0–100 | 98 a | 91.0–100 | 98.0 b |
| NSP2 | 66.0–100 | 86.0 a | 82.0–100 | 89.0 ab | 93.0–100 | 99.0 b |
| NSP3 | 58.0–100 | 100 b | 94.0–100 | 99 a | 89.0–100 | 98.0 a |
| NSP4 | 77.0–100 | 93.0 b | 96.0–100 | 99.0 a | 90.0–100 | 98.0 a |
| NSP5 | 44.0–100 | 81.0 b | 77.0–100 | 94.0 a | 89.0–100 | 97.0 c |
| Gene Segment | Number of Sequences | Previously Proposed Nucleotide Cutoff | Reference | Currently Proposed Nucleotide Cutoff | Genotypes in Each Host Species | ||||
|---|---|---|---|---|---|---|---|---|---|
| Murine | Human | Bovine | Caprine | Porcine | |||||
| VP7 | 419 | 80% | Marthaler et al., 2012 | 80% | G1 | G2 | G3, G5 | G3 | G4, G6–G26 |
| VP4 | 64 | -- | -- | 80% | P[1] | P[2] | P[3] | P[3] | P[4], P[5] |
| VP6 | 144 | 81% | Marthaler et al., 2014 | 81% | I1 | I2 | I3 | I3 | I4–I13 |
| VP1 | 54 | -- | -- | 78% | R1 | R2 | R5 | R3 | R4 |
| VP2 | 57 | -- | -- | 79% | C1 | C2 | C3, C5 | C3 | C4 |
| VP3 | 61 | 70% | Hayashi-Miyamoto et al., 2017 | 77% * | M1 | M2 | M3 | M3 | M4, M5 |
| NSP1 | 68 | 76% | Suzuki et al., 2011 | 76% | A1 | A2 | A4, A5 | A3, A4 | A6–A8 |
| NSP2 | 89 | 75% | Suzuki et al., 2012 | 83% * | N1 | N2 | N3, N4 | N3 | N5–N10 |
| NSP3 | 58 | 78% | Hayashi-Miyamoto et al., 2017 | 78% | T1 | T2 | T3 | T3 | T4–T6 |
| NSP4 | 68 | 70% | Hayashi-Miyamoto et al., 2017 | 76% * | E1 | E2 | E3 | E3 | E4 |
| NSP5 | 95 | 78% | Suzuki et al., 2012 | 79% * | H1 | H2 | H3, H5 | H3 | H4, H6, H7 |
| VP7 | VP4 | VP6 | VP1 | VP2 | VP3 | NSP1 | NSP2 | NSP3 | NSP4 | NSP5 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| RVB/Rat-wt/USA/IDIR/XXXX | G1 | P[1] | I1 | R1 | C1 | M1 | A1 | N1 | T1 | E1 | H1 |
| RVB/Human-wt ^ | G2 | P[2] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| RVB/Bovine-wt/IND/DB176/2001 | G5 | P[3] | I3 | R5 | C5 | na | A5 | N4 | na | na | H5 |
| RVB/Bovine-wt/IND/RUBV226/2004 | G5 | P[3] | I3 | R5 | C5 | na | A5 | N4 | na | na | na |
| RVB/Bovine-wt/IND/RUBV282/2005 | G5 | P[3] | I3 | R5 | C5 | na | A5 | N4 | na | na | H5 |
| RVB/Bovine-wt/JPN/G-2006/2006/G3PX | G3 | P[3] | na | na | na | M3 | A3 | N3 | T3 | E3 | H3 |
| RVB/Bovine-wt/JPN/IS-1/1999/G3PX | G3 | P[3] | * | * | na | M3 | A3 | * | T3 | E3 | na |
| RVB/Bovine-wt/JPN/IS-2/2002/G3PX | G3 | P[3] | * | * | C3 | M3 | na | na | T3 | E3 | na |
| RVB/Goat-wt/USA/CA22/2014 | G3 | P[3] | I3 | na | C3 | na | A3 | N3 | T3 | E3 | H3 |
| RVB/Goat-wt/USA/Minnesota-1/2016 | G3 | P[3] | I3 | R3 | C3 | M3 | A3 | N3 | T3 | E3 | H3 |
| RVB/Pig-tc/USA/LS00011_Ohio/XXXX/GXP[X] | G12 | P[4] | I13 | R4 | C4 | M4 | A8 | N10 | T6 | E4 | H7 |
| RVB/Pig-wt/USA/IL10/2013 & IL10B/2013 | G16 | P[4]/P[5] | I13 | R4 | C4 | M4 | A8 | N10 | T4 | E4 | H7 |
| RVB/Pig-wt/USA/IL11/2013 | G16 | P[4] | I13 | R4 | C4 | M4 | A8 | N10 | T4 | E4 | H7 |
| RVB/Pig-wt/USA/IL12/2013 & IL12B/2013 | G16 | P[4]/P[5] | I13 | R4 | C4 | M4 | A8 | N10 | T4 | E4 | H7 |
| RVB/Pig-wt/USA/IL13/2013 | G16 | P[4] | I13 | R4 | C4 | M4 | A8 | N10 | T4 | E4 | H7 |
| RVB/Pig-wt/USA/IL14/2013 & IL14B/2013 | G16 | P[4]/P[5] | I13 | R4 | C4 | M4 | A8 | N10 | T4 | E4 | H7 |
| RVB/Pig-wt/USA/IL15/2013 & L15B/2013 | G16 | P[4]/P[5] | I13 | R4 | C4 | M4 | A8 | N10 | T4 | E4 | H7 |
| RVB/Pig-wt/USA/IL16/2013 | G16 | P[5] | I13 | R4 | C4 | M4 | A8 | N10 | T4 | E4 | H7 |
| RVB/Pig-wt/USA/IL17/2013 | G16 | P[5] | I13 | R4 | C4 | M4 | A8 | N10 | T4 | E4 | H7 |
| RVB/Pig-wt/USA/IL18/2013 | G16 | P[5] | I13 | R4 | C4 | M4 | A8 | N10 | T4 | E4 | H7 |
| RVB/Pig-wt/USA/IL19/2013 | G16 | P[5] | I13 | R4 | C4 | M4 | A8 | N10 | T4 | E4 | H7 |
| RVB/Pig-wt/USA/IL20/2013 | G16 | P[5] | I13 | R4 | C4 | M4 | A8 | N10 | T4 | E4 | H7 |
| RVB/Pig-wt/USA/IL21/2013 | G16 | P[5] | I13 | R4 | C4 | M4 | A8 | N10 | T4 | E4 | H7 |
| RVB/Pig-wt/USA/IL3/2012 | G16 | P[5] | I13 | R4 | C4 | M4 | A8 | N10 | T4 | E4 | H7 |
| RVB/Pig-wt/USA/IL4/2012 | G16 | P[5] | I13 | R4 | C4 | M4 | A8 | N10 | T4 | E4 | H7 |
| RVB/Pig-wt/USA/IL5/2012 | G16 | P[4] | I13 | R4 | C4 | M4 | A8 | N10 | T4 | E4 | H7 |
| RVB/Pig-wt/USA/IL6/2012 | G16 | P[5] | I13 | R4 | C4 | M4 | A8 | N10 | T4 | E4 | H7 |
| RVB/Pig-wt/USA/IL7/2012 | G16 | P[4] | I13 | R4 | C4 | M4 | A8 | N10 | T4 | E4 | H7 |
| RVB/Pig-wt/USA/IL8/2013 & IL8B/2013 | G16 | P[4]/P[5] | I13 | R4 | C4 | M4 | A8 | N10 | T4 | E4 | H7 |
| RVB/Pig-wt/USA/IL9/2013 | G16 | P[5] | I13 | R4 | C4 | M4 | A8 | N10 | T4 | E4 | H7 |
| RVB/Pig-wt/USA/KS1/2012 | G16 | P[4] | I13 | R4 | C4 | M4 | A8 | N10 | T5 | E4 | H7 |
| RVB/Pig-wt/USA/KS2/2012 & KS2B/2012 | G14/G16 | na | I13 | R4 | na | M4 | A8 | N10 | T5 | E4 | H7 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shepherd, F.K.; Herrera-Ibata, D.M.; Porter, E.; Homwong, N.; Hesse, R.; Bai, J.; Marthaler, D.G. Whole Genome Classification and Phylogenetic Analyses of Rotavirus B strains from the United States. Pathogens 2018, 7, 44. https://doi.org/10.3390/pathogens7020044
Shepherd FK, Herrera-Ibata DM, Porter E, Homwong N, Hesse R, Bai J, Marthaler DG. Whole Genome Classification and Phylogenetic Analyses of Rotavirus B strains from the United States. Pathogens. 2018; 7(2):44. https://doi.org/10.3390/pathogens7020044
Chicago/Turabian StyleShepherd, Frances K., Diana Maria Herrera-Ibata, Elizabeth Porter, Nitipong Homwong, Richard Hesse, Jianfa Bai, and Douglas G. Marthaler. 2018. "Whole Genome Classification and Phylogenetic Analyses of Rotavirus B strains from the United States" Pathogens 7, no. 2: 44. https://doi.org/10.3390/pathogens7020044
APA StyleShepherd, F. K., Herrera-Ibata, D. M., Porter, E., Homwong, N., Hesse, R., Bai, J., & Marthaler, D. G. (2018). Whole Genome Classification and Phylogenetic Analyses of Rotavirus B strains from the United States. Pathogens, 7(2), 44. https://doi.org/10.3390/pathogens7020044