The Cooperative Functions of the EBNA3 Proteins Are Central to EBV Persistence and Latency


{kind=link}
{kind=link}
Abstract
1. Epstein–Barr Virus
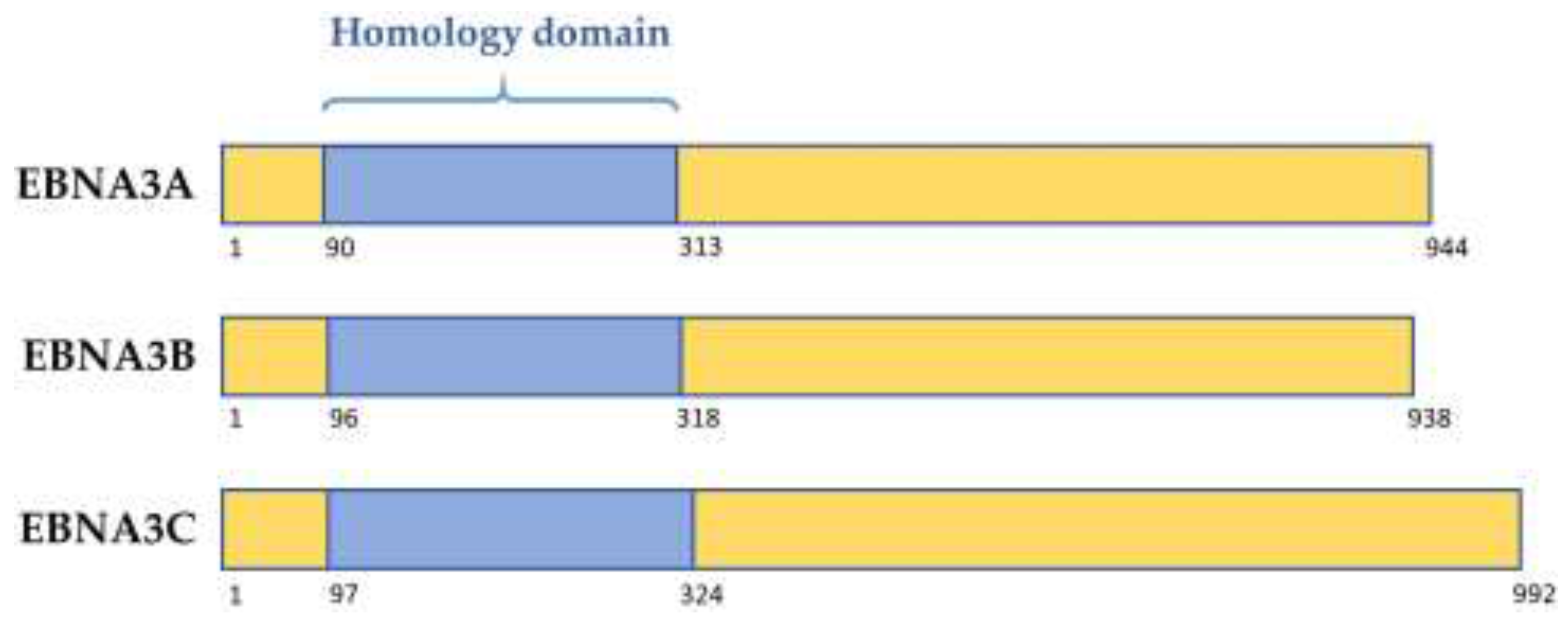
2. The EBNA3 Family
3. EBNA3 Regulatory Mechanisms
3.1. Recruitment
3.2. Changes in Chromatin Architecture
3.3. Repressors and Activators
3.4. EBNA3 Interactors That Facilitate Regulation
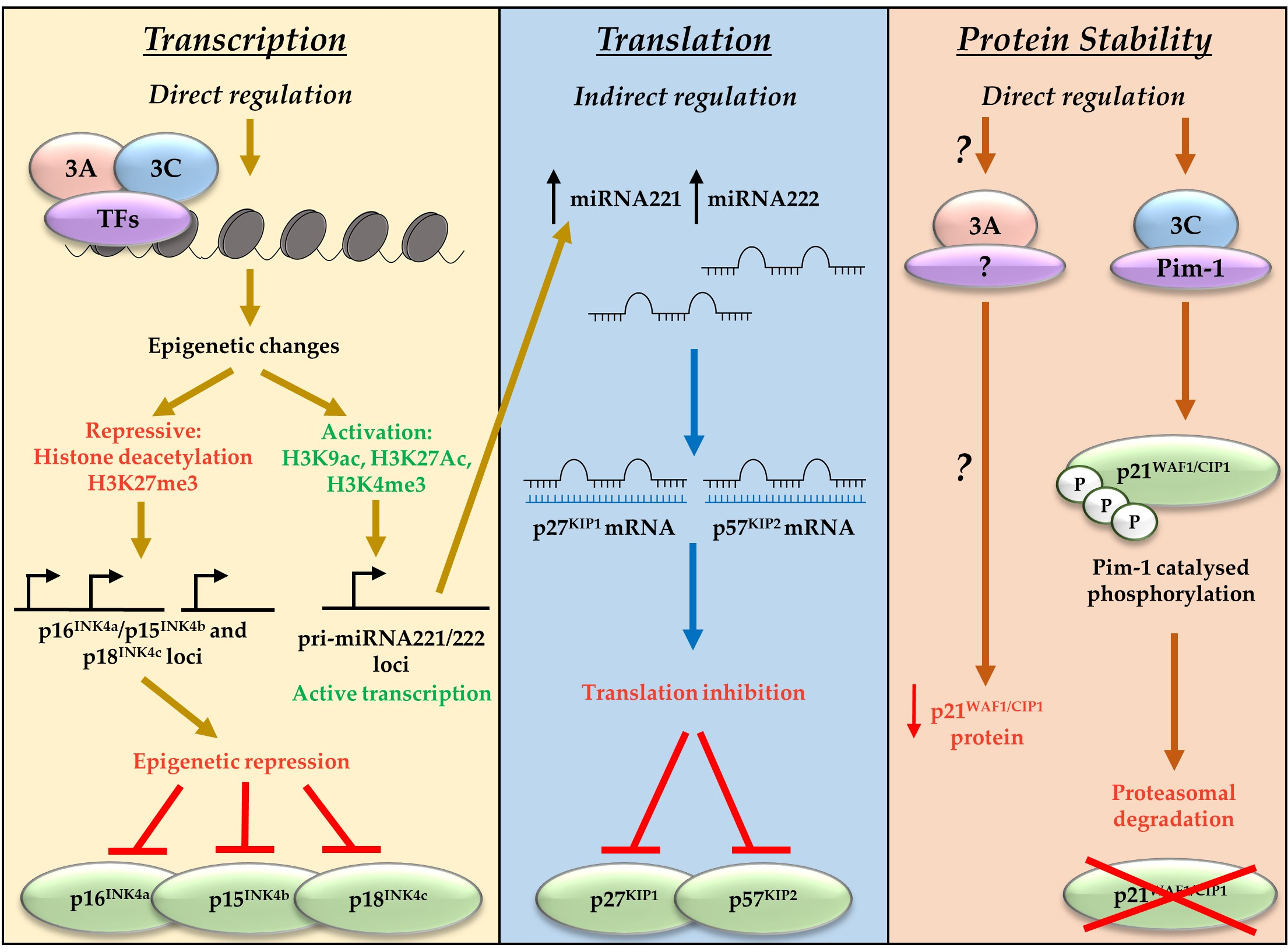
4. EBNA3A and EBNA3C Regulate Anti-Proliferative and Tumour Suppressive Pathways
4.1. Direct Epigenetic Repression of the Tumour Suppressor and CDKI p16INK4a
4.2. EBNA3A- and EBNA3C-Mediated Transactivation of Oncogenic miRNAs That Target CDKIs
5. Anti-Apoptotic Functions of EBNA3A and EBNA3C
6. EBNA3B: The Antagonistic Tumour Suppressive EBNA3 Protein
7. Suppression of Plasma Cell Differentiation: An Additional Role for the Oncogenic EBNA3 Proteins
8. Summary
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Klein, G. Perspectives in studies of human tumor viruses. Front. Biosci. 2002, 7, d268–d274. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S.; Rickinson, A.B. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 2004, 4, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Roughan, J.E.; Thorley-Lawson, D.A. The intersection of Epstein-Barr virus with the germinal center. J. Virol. 2009, 83, 3968–3976. [Google Scholar] [CrossRef] [PubMed]
- Yenamandra, S.P.; Sompallae, R.; Klein, G.; Kashuba, E. Comparative analysis of the Epstein-Barr virus encoded nuclear proteins of EBNA-3 family. Comput. Biol. Med. 2009, 39, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Le Roux, A.; Kerdiles, B.; Walls, D.; Dedieu, J.F.; Perricaudet, M. The Epstein-Barr virus determined nuclear antigens EBNA-3A, -3B, and -3C repress EBNA-2-mediated transactivation of the viral terminal protein 1 gene promoter. Virology 1994, 205, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Touitou, R.; O’Nions, J.; Heaney, J.; Allday, M.J. Epstein-Barr virus EBNA3 proteins bind to the c8/alpha7 subunit of the 20s proteasome and are degraded by 20s proteasomes in vitro, but are very stable in latently infected B cells. J. Gen. Virol. 2005, 86, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.L.; Xu, S.; Lyons-Weiler, J.; Rosendorff, A.; Webber, S.A.; Wasil, L.R.; Metes, D.; Rowe, D.T. Cellular factors associated with latency and spontaneous Epstein-Barr virus reactivation in B-lymphoblastoid cell lines. Virology 2010, 400, 53–67. [Google Scholar] [CrossRef] [PubMed]
- White, R.E.; Groves, I.J.; Turro, E.; Yee, J.; Kremmer, E.; Allday, M.J. Extensive co-operation between the Epstein-Barr virus EBNA3 proteins in the manipulation of host gene expression and epigenetic chromatin modification. PLoS ONE 2010, 5, e13979. [Google Scholar] [CrossRef] [PubMed]
- Anderton, E.; Yee, J.; Smith, P.; Crook, T.; White, R.E.; Allday, M.J. Two Epstein-Barr virus (EBV) oncoproteins cooperate to repress expression of the proapoptotic tumour-suppressor BIM: Clues to the pathogenesis of Burkitt’s lymphoma. Oncogene 2008, 27, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Skalska, L.; White, R.E.; Franz, M.; Ruhmann, M.; Allday, M.J. Epigenetic repression of p16(INK4a) by latent Epstein-Barr virus requires the interaction of EBNA3A and EBNA3C with CtBP. PLoS Pathog. 2010, 6, e1000951. [Google Scholar] [CrossRef] [PubMed]
- Maruo, S.; Wu, Y.; Ishikawa, S.; Kanda, T.; Iwakiri, D.; Takada, K. Epstein-Barr virus nuclear protein EBNA3C is required for cell cycle progression and growth maintenance of lymphoblastoid cells. Proc. Natl. Acad. Sci. USA 2006, 103, 19500–19505. [Google Scholar] [CrossRef] [PubMed]
- Bazot, Q.; Paschos, K.; Skalska, L.; Kalchschmidt, J.S.; Parker, G.A.; Allday, M.J. Epstein-Barr virus proteins EBNA3A and EBNA3C together induce expression of the oncogenic microRNA cluster miR-221/miR-222 and ablate expression of its target p57KIP2. PLoS Pathog. 2015, 11, e1005031. [Google Scholar] [CrossRef] [PubMed]
- Styles, C.T.; Bazot, Q.; Parker, G.A.; White, R.E.; Paschos, K.; Allday, M.J. EBV epigenetically suppresses the B cell-to-plasma cell differentiation pathway while establishing long-term latency. PLoS Biol. 2017, 15, e2001992. [Google Scholar] [CrossRef] [PubMed]
- Maruo, S.; Johannsen, E.; Illanes, D.; Cooper, A.; Kieff, E. Epstein-Barr virus nuclear protein EBNA3A is critical for maintaining lymphoblastoid cell line growth. J. Virol. 2003, 77, 10437–10447. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Paschos, K.; Parker, G.A.; Watanatanasup, E.; White, R.E.; Allday, M.J. BIM promoter directly targeted by EBNA3C in polycomb-mediated repression by EBV. Nucleic Acids Res. 2012, 40, 7233–7246. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Willox, B.; Zhou, H.; Holthaus, A.M.; Wang, A.; Shi, T.T.; Maruo, S.; Kharchenko, P.V.; Johannsen, E.C.; Kieff, E.; et al. Epstein-Barr virus nuclear antigen 3C binds to BATF/IRF4 or SPI1/IRF4 composite sites and recruits SIN3A to repress CDKN2A. Proc. Natl. Acad. Sci. USA 2014, 111, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.C.; Jiang, S.; Zhou, H.; Willox, B.; Holthaus, A.M.; Kharchenko, P.V.; Johannsen, E.C.; Kieff, E.; Zhao, B. Epstein-Barr virus nuclear antigen 3A partially coincides with EBNA3C genome-wide and is tethered to DNA through BATF complexes. Proc. Natl. Acad. Sci. USA 2015, 112, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Paschos, K.; Bazot, Q.; Ho, G.; Parker, G.A.; Lees, J.; Barton, G.; Allday, M.J. Core binding factor (CBF) is required for Epstein-Barr virus EBNA3 proteins to regulate target gene expression. Nucl. Acids Res. 2016, 45, 2368–2383. [Google Scholar] [CrossRef] [PubMed]
- Parker, G.A.; Crook, T.; Bain, M.; Sara, E.A.; Farrell, P.J.; Allday, M.J. Epstein-Barr virus nuclear antigen (EBNA)3C is an immortalizing oncoprotein with similar properties to adenovirus E1A and papillomavirus E7. Oncogene 1996, 13, 2541–2549. [Google Scholar] [PubMed]
- Allday, M.J.; Bazot, Q.; White, R.E. The EBNA3 family: Two oncoproteins and a tumour suppressor that are central to the biology of EBV in B cells. Curr. Top. Microbiol. Immunol. 2015, 391, 61–117. [Google Scholar] [PubMed]
- Lee, M.A.; Diamond, M.E.; Yates, J.L. Genetic evidence that EBNA-1 is needed for efficient, stable latent infection by Epstein-Barr virus. J. Virol. 1999, 73, 2974–2982. [Google Scholar] [PubMed]
- Cohen, J.I.; Wang, F.; Mannick, J.; Kieff, E. Epstein-Barr virus nuclear protein 2 is a key determinant of lymphocyte transformation. Proc. Natl. Acad. Sci. USA 1989, 86, 9558–9562. [Google Scholar] [CrossRef] [PubMed]
- Tomkinson, B.; Robertson, E.; Kieff, E. Epstein-Barr virus nuclear proteins EBNA-3A and EBNA-3C are essential for B-lymphocyte growth transformation. J. Virol. 1993, 67, 2014–2025. [Google Scholar] [PubMed]
- Kaye, K.M.; Izumi, K.M.; Kieff, E. Epstein-Barr virus latent membrane protein 1 is essential for B-lymphocyte growth transformation. Proc. Natl. Acad. Sci. USA 1993, 90, 9150–9154. [Google Scholar] [CrossRef] [PubMed]
- Mannick, J.B.; Cohen, J.I.; Birkenbach, M.; Marchini, A.; Kieff, E. The Epstein-Barr virus nuclear protein encoded by the leader of the EBNA RNAs is important in B-lymphocyte transformation. J. Virol. 1991, 65, 6826–6837. [Google Scholar] [PubMed]
- Hertle, M.L.; Popp, C.; Petermann, S.; Maier, S.; Kremmer, E.; Lang, R.; Mages, J.; Kempkes, B. Differential gene expression patterns of EBV infected EBNA-3A positive and negative human b lymphocytes. PLoS Pathog. 2009, 5, e1000506. [Google Scholar] [CrossRef] [PubMed]
- White, R.E.; Rämer, P.C.; Naresh, K.N.; Meixlsperger, S.; Pinaud, L.; Rooney, C.; Savoldo, B.; Coutinho, R.; Bödör, C.; Gribben, J.; et al. EBNA3B-deficient EBV promotes B cell lymphomagenesis in humanized mice and is found in human tumors. J. Clin. Invest. 2012, 122, 1487–1502. [Google Scholar] [CrossRef] [PubMed]
- Robertson, E.S.; Lin, J.; Kieff, E. The amino-terminal domains of Epstein-Barr virus nuclear proteins 3A, 3B, and 3C interact with RBPJ(kappa). J. Virol. 1996, 70, 3068–3074. [Google Scholar] [PubMed]
- Robertson, E.S.; Grossman, S.; Johannsen, E.; Miller, C.; Lin, J.; Tomkinson, B.; Kieff, E. Epstein-Barr virus nuclear protein 3C modulates transcription through interaction with the sequence-specific DNA-binding protein J kappa. J. Virol. 1995, 69, 3108–3116. [Google Scholar] [PubMed]
- Kalchschmidt, J.S.; Gillman, A.C.; Paschos, K.; Bazot, Q.; Kempkes, B.; Allday, M.J. EBNA3C directs recruitment of RBPJ (CBF1) to chromatin during the process of gene repression in EBV infected B cells. PLoS Pathog. 2016, 12, e1005383. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Marshall, D.R.; Sample, C.E. A conserved domain of the Epstein-Barr virus nuclear antigens 3A and 3C binds to a discrete domain of J kappa. J. Virol. 1996, 70, 4228–4236. [Google Scholar] [PubMed]
- West, M.J. Chromatin reorganisation in Epstein-Barr virus-infected cells and its role in cancer development. Curr. Opin. Virol. 2017, 26, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Johannsen, E.; Miller, C.L.; Grossman, S.R.; Kieff, E. EBNA-2 and EBNA-3C extensively and mutually exclusively associate with RBPJkappa in Epstein-Barr virus-transformed B lymphocytes. J. Virol. 1996, 70, 4179–4183. [Google Scholar] [PubMed]
- Radkov, S.A.; Bain, M.; Farrell, P.J.; West, M.; Rowe, M.; Allday, M.J. Epstein-Barr virus EBNA3C represses Cp, the major promoter for EBNA expression, but has no effect on the promoter of the cell gene CD21. J. Virol. 1997, 71, 8552–8562. [Google Scholar] [PubMed]
- Maruo, S.; Johannsen, E.; Illanes, D.; Cooper, A.; Zhao, B.; Kieff, E. Epstein-Barr virus nuclear protein 3A domains essential for growth of lymphoblasts: Transcriptional regulation through RBP-jkappa/CBF1 is critical. J. Virol. 2005, 79, 10171–10179. [Google Scholar] [CrossRef] [PubMed]
- Maruo, S.; Wu, Y.; Ito, T.; Kanda, T.; Kieff, E.D.; Takada, K. Epstein-Barr virus nuclear protein EBNA3C residues critical for maintaining lymphoblastoid cell growth. Proc. Natl. Acad. Sci. USA 2009, 106, 4419–4424. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Sakakibara, S.; Maruo, S.; Zhao, B.; Calderwood, M.A.; Holthaus, A.M.; Lai, C.Y.; Takada, K.; Kieff, E.; Johannsen, E. Epstein-Barr virus nuclear protein 3C domains necessary for lymphoblastoid cell growth: Interaction with RBP-Jkappa regulates TCL1. J. Virol. 2009, 83, 12368–12377. [Google Scholar] [CrossRef] [PubMed]
- McClellan, M.J.; Wood, C.D.; Ojeniyi, O.; Cooper, T.J.; Kanhere, A.; Arvey, A.; Webb, H.M.; Palermo, R.D.; Harth-Hertle, M.L.; Kempkes, B.; et al. Modulation of enhancer looping and differential gene targeting by Epstein-Barr virus transcription factors directs cellular reprogramming. PLoS Pathog. 2013, 9, e1003636. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Welch, R.; Zhao, B.; Ta, T.; Keles, S.; Johannsen, E. Epstein-Barr virus nuclear antigen 3 (EBNA3) proteins regulate EBNA2 binding to distinct RBPJ genomic sites. J. Virol. 2015, 90, 2906–2919. [Google Scholar] [CrossRef] [PubMed]
- Harth-Hertle, M.L.; Scholz, B.A.; Erhard, F.; Glaser, L.V.; Dölken, L.; Zimmer, R.; Kempkes, B. Inactivation of intergenic enhancers by EBNA3A initiates and maintains polycomb signatures across a chromatin domain encoding CXCL10 and CXCL9. PLoS Pathog. 2013, 9, e1003638. [Google Scholar] [CrossRef] [PubMed]
- Wood, C.D.; Veenstra, H.; Khasnis, S.; Gunnell, A.; Webb, H.M.; Shannon-Lowe, C.; Andrews, S.; Osborne, C.S.; West, M.J. MYC activation and BCL2L11 silencing by a tumour virus through the large-scale reconfiguration of enhancer-promoter hubs. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Skalska, L.; White, R.E.; Parker, G.A.; Turro, E.; Sinclair, A.J.; Paschos, K.; Allday, M.J. Induction of p16(INK4a) is the major Barrier to proliferation when Epstein-Barr virus (EBV) transforms primary B cells into lymphoblastoid cell lines. PLoS Pathog. 2013, 9, e1003187. [Google Scholar] [CrossRef]
- Hickabottom, M.; Parker, G.A.; Freemont, P.; Crook, T.; Allday, M.J. Two nonconsensus sites in the Epstein-Barr virus oncoprotein EBNA3A cooperate to bind the co-repressor carboxyl-terminal-binding protein (CtBP). J. Biol. Chem. 2002, 277, 47197–47204. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.S.; Lan, K.; Subramanian, C.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C recruits histone deacetylase activity and associates with the corepressors mSIN3A and NCoR in human B-cell lines. J. Virol. 2003, 77, 4261–4272. [Google Scholar] [CrossRef] [PubMed]
- Radkov, S.A.; Touitou, R.; Brehm, A.; Rowe, M.; West, M.; Kouzarides, T.; Allday, M.J. Epstein-Barr virus nuclear antigen 3C interacts with histone deacetylase to repress transcription. J. Virol. 1999, 73, 5688–5697. [Google Scholar] [PubMed]
- Touitou, R.; Hickabottom, M.; Parker, G.; Crook, T.; Allday, M.J. Physical and functional interactions between the corepressor CtBP and the Epstein-Barr virus nuclear antigen EBNA3C. J. Virol. 2001, 75, 7749–7755. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cotter, M.A., II; Robertson, E.S. Modulation of histone acetyltransferase activity through interaction of Epstein-Barr nuclear antigen 3C with prothymosin alpha. Mol. Cell. Biol. 2000, 20, 5722–5735. [Google Scholar] [CrossRef] [PubMed]
- Kalchschmidt, J.S.; Bashford-Rogers, R.; Paschos, K.; Gillman, A.C.; Styles, C.T.; Kellam, P.; Allday, M.J. Epstein-Barr virus nuclear protein EBNA3C directly induces expression of AID and somatic mutations in B cells. J. Exp. Med. 2016, 213, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Frangini, A.; Sjoberg, M.; Roman-Trufero, M.; Dharmalingam, G.; Haberle, V.; Bartke, T.; Lenhard, B.; Malumbres, M.; Vidal, M.; Dillon, N. The aurora B kinase and the polycomb protein Ring1b combine to regulate active promoters in quiescent lymphocytes. Mol. Cell 2013, 51, 647–661. [Google Scholar] [CrossRef] [PubMed]
- McClellan, M.J.; Khasnis, S.; Wood, C.D.; Palermo, R.D.; Schlick, S.N.; Kanhere, A.S.; Jenner, R.G.; West, M.J. Downregulation of integrin receptor-signaling genes by Epstein-Barr virus EBNA 3C via promoter-proximal and -distal binding elements. J. Virol. 2012, 86, 5165–5178. [Google Scholar] [CrossRef] [PubMed]
- Kuzmichev, A.; Nishioka, K.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Histone methyltransferase activity associated with a human multiprotein complex containing the enhancer of zeste protein. Genes Dev. 2002, 16, 2893–2905. [Google Scholar] [CrossRef] [PubMed]
- Bazot, Q.; Paschos, K.; Allday, M.J. Epstein-Barr virus latent protein EBNA3A directly targets and silences the kinase STK39 in B cells infected by EBV. J. Virol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Allday, M.J. EBV finds a polycomb-mediated, epigenetic solution to the problem of oncogenic stress responses triggered by infection. Front. Genet. 2013, 4, 212. [Google Scholar] [CrossRef] [PubMed]
- Maruo, S.; Zhao, B.; Johannsen, E.; Kieff, E.; Zou, J.; Takada, K. Epstein-Barr virus nuclear antigens 3C and 3A maintain lymphoblastoid cell growth by repressing p16INK4a and p14ARF expression. Proc. Natl. Acad. Sci. USA 2011, 108, 1919–1924. [Google Scholar] [CrossRef] [PubMed]
- Popov, N.; Gil, J. Epigenetic regulation of the INK4b-ARF-INK4a locus: In sickness and in health. Epigenetics 2010, 5, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Chinnadurai, G. The transcriptional corepressor CtBP: A foe of multiple tumor suppressors. Cancer Res. 2009, 69, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Bazot, Q.; Deschamps, T.; Tafforeau, L.; Siouda, M.; Leblanc, P.; Harth-Hertle, M.L.; Rabourdin-Combe, C.; Lotteau, V.; Kempkes, B.; Tommasino, M.; et al. Epstein-Barr virus nuclear antigen 3A protein regulates CDKN2B transcription via interaction with MIZ-1. Nucleic Acids Res. 2014, 42, 9700–9716. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Zhou, H.; Liang, J.; Gerdt, C.; Wang, C.; Ke, L.; Schmidt, S.C.S.; Narita, Y.; Ma, Y.; Wang, S.; et al. The Epstein-Barr virus regulome in lymphoblastoid cells. Cell Host Microbe 2017, 22, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Lu, J.; Cai, Q.; Sun, Z.; Jha, H.C.; Robertson, E.S. EBNA3C augments Pim-1 mediated phosphorylation and degradation of p21 to promote B-cell proliferation. PLoS Pathog. 2014, 10, e1004304. [Google Scholar] [CrossRef] [PubMed]
- Tursiella, M.L.; Bowman, E.R.; Wanzeck, K.C.; Throm, R.E.; Liao, J.; Zhu, J.; Sample, C.E. Epstein-Barr virus nuclear antigen 3A promotes cellular proliferation by repression of the cyclin-dependent kinase inhibitor p21WAF1/CIP1. PLoS Pathog. 2014, 10, e1004415. [Google Scholar] [CrossRef] [PubMed]
- Strasser, A. The role of BH3-only proteins in the immune system. Nat. Rev. Immunol. 2005, 5, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Enders, A.; Bouillet, P.; Puthalakath, H.; Xu, Y.; Tarlinton, D.M.; Strasser, A. Loss of the pro-apoptotic BH3-only BCL-2 family member BIM inhibits BCR stimulation-induced apoptosis and deletion of autoreactive B cells. J. Exp. Med. 2003, 198, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.F.; Bouillet, P.; O’Donnell, K.; Light, A.; Tarlinton, D.M.; Strasser, A. Proapoptotic BH3-only protein BIM is essential for developmentally programmed death of germinal center-derived memory B cells and antibody-forming cells. Blood 2007, 110, 3978–3984. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; O’Donnell K, A.; Juopperi, T. The great MYC escape in tumorigenesis. Cancer Cell 2005, 8, 177–178. [Google Scholar] [CrossRef] [PubMed]
- Egle, A.; Harris, A.W.; Bouillet, P.; Cory, S. BIM is a suppressor of MYC-induced mouse B cell leukemia. Proc. Natl. Acad. Sci. USA 2004, 101, 6164–6169. [Google Scholar] [CrossRef] [PubMed]
- Paschos, K.; Smith, P.; Anderton, E.; Middeldorp, J.M.; White, R.E.; Allday, M.J. Epstein-Barr virus latency in B cells leads to epigenetic repression and CpG methylation of the tumour suppressor gene BIM. PLoS Pathog. 2009, 5, e1000492. [Google Scholar] [CrossRef] [PubMed]
- Thorley-Lawson, D.A.; Allday, M.J. The curious case of the tumour virus: 50 years of Burkitt’s lymphoma. Nat. Rev. Microbiol. 2008, 6, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Nikitin, P.A.; Yan, C.M.; Forte, E.; Bocedi, A.; Tourigny, J.P.; White, R.E.; Allday, M.J.; Patel, A.; Dave, S.S.; Kim, W.; et al. An ATM/CHK2-mediated DNA damage-responsive signaling pathway suppresses Epstein-Barr virus transformation of primary human B cells. Cell Host Microbe 2010, 8, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Murakami, M.; Kumar, P.; Bajaj, B.; Sims, K.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C augments MDM2-mediated p53 ubiquitination and degradation by deubiquitinating MDM2. J. Virol. 2009, 83, 4652–4669. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; Saha, A.; Murakami, M.; Kumar, P.; Knight, J.S.; Cai, Q.; Choudhuri, T.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C targets p53 and modulates its transcriptional and apoptotic activities. Virology 2009, 388, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Jha, H.C.; A, J.M.; Saha, A.; Banerjee, S.; Lu, J.; Robertson, E.S. Epstein-Barr virus essential antigen EBNA3C attenuates H2AX expression. J. Virol. 2014, 88, 3776–3788. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.S.; Sharma, N.; Robertson, E.S. Epstein-Barr virus latent antigen 3C can mediate the degradation of the retinoblastoma protein through an SCF cellular ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2005, 102, 18562–18566. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Lu, J.; Morizur, L.; Upadhyay, S.K.; Aj, M.P.; Robertson, E.S. E2F1 mediated apoptosis induced by the DNA damage response is blocked by EBV nuclear antigen 3C in lymphoblastoid cells. PLoS Pathog. 2012, 8, e1002573. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.S.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C regulates cyclin a/p27 complexes and enhances cyclin a-dependent kinase activity. J. Virol. 2004, 78, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.S.; Sharma, N.; Robertson, E.S. Scfskp2 complex targeted by Epstein-Barr virus essential nuclear antigen. Mol. Cell. Biol. 2005, 25, 1749–1763. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, B.G.; Murakami, M.; Cai, Q.; Verma, S.C.; Lan, K.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C interacts with and enhances the stability of the c-MYC oncoprotein. J. Virol. 2008, 82, 4082–4090. [Google Scholar] [CrossRef] [PubMed]
- Tomkinson, B.; Kieff, E. Use of second-site homologous recombination to demonstrate that Epstein-Barr virus nuclear protein 3B is not important for lymphocyte infection or growth transformation in vitro. J. Virol. 1992, 66, 2893–2903. [Google Scholar] [PubMed]
- Gagrica, S.; Brookes, S.; Anderton, E.; Rowe, J.; Peters, G. Contrasting behavior of the p18INK4c and p16INK4a tumor suppressors in both replicative and oncogene-induced senescence. Cancer Res. 2012, 72, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Morse, L.; Chen, D.; Franklin, D.; Xiong, Y.; Chen-Kiang, S. Induction of cell cycle arrest and B cell terminal differentiation by cdk inhibitor p18(INK4c) and il-6. Immunity 1997, 6, 47–56. [Google Scholar] [CrossRef]
- Tourigny, M.R.; Ursini-Siegel, J.; Lee, H.; Toellner, K.M.; Cunningham, A.F.; Franklin, D.S.; Ely, S.; Chen, M.; Qin, X.F.; Xiong, Y.; et al. Cdk inhibitor p18(INK4c) is required for the generation of functional plasma cells. Immunity 2002, 17, 179–189. [Google Scholar] [CrossRef]
- Boi, M.; Zucca, E.; Inghirami, G.; Bertoni, F. PRDM1/BLIMP1: A tumor suppressor gene in B and T cell lymphomas. Leuk. Lymphoma 2015, 56, 1223–1228. [Google Scholar] [CrossRef] [PubMed]
- Mandelbaum, J.; Bhagat, G.; Tang, H.; Mo, T.; Brahmachary, M.; Shen, Q.; Chadburn, A.; Rajewsky, K.; Tarakhovsky, A.; Pasqualucci, L.; et al. BLIMP1 is a tumor suppressor gene frequently disrupted in activated B cell-like diffuse large B cell lymphoma. Cancer Cell 2010, 18, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Minnich, M.; Tagoh, H.; Bönelt, P.; Axelsson, E.; Fischer, M.; Cebolla, B.; Tarakhovsky, A.; Nutt, S.L.; Jaritz, M.; Busslinger, M. Multifunctional role of the transcription factor BLIMP-1 in coordinating plasma cell differentiation. Nat. Immunol. 2016, 17, 331–343. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Styles, C.T.; Paschos, K.; White, R.E.; Farrell, P.J. The Cooperative Functions of the EBNA3 Proteins Are Central to EBV Persistence and Latency. Pathogens 2018, 7, 31. https://doi.org/10.3390/pathogens7010031
Styles CT, Paschos K, White RE, Farrell PJ. The Cooperative Functions of the EBNA3 Proteins Are Central to EBV Persistence and Latency. Pathogens. 2018; 7(1):31. https://doi.org/10.3390/pathogens7010031
Chicago/Turabian StyleStyles, Christine T., Kostas Paschos, Robert E. White, and Paul J. Farrell. 2018. "The Cooperative Functions of the EBNA3 Proteins Are Central to EBV Persistence and Latency" Pathogens 7, no. 1: 31. https://doi.org/10.3390/pathogens7010031
APA StyleStyles, C. T., Paschos, K., White, R. E., & Farrell, P. J. (2018). The Cooperative Functions of the EBNA3 Proteins Are Central to EBV Persistence and Latency. Pathogens, 7(1), 31. https://doi.org/10.3390/pathogens7010031