Longitudinal Surveillance of Porcine Rotavirus B Strains from the United States and Canada and In Silico Identification of Antigenically Important Sites


Abstract
1. Introduction
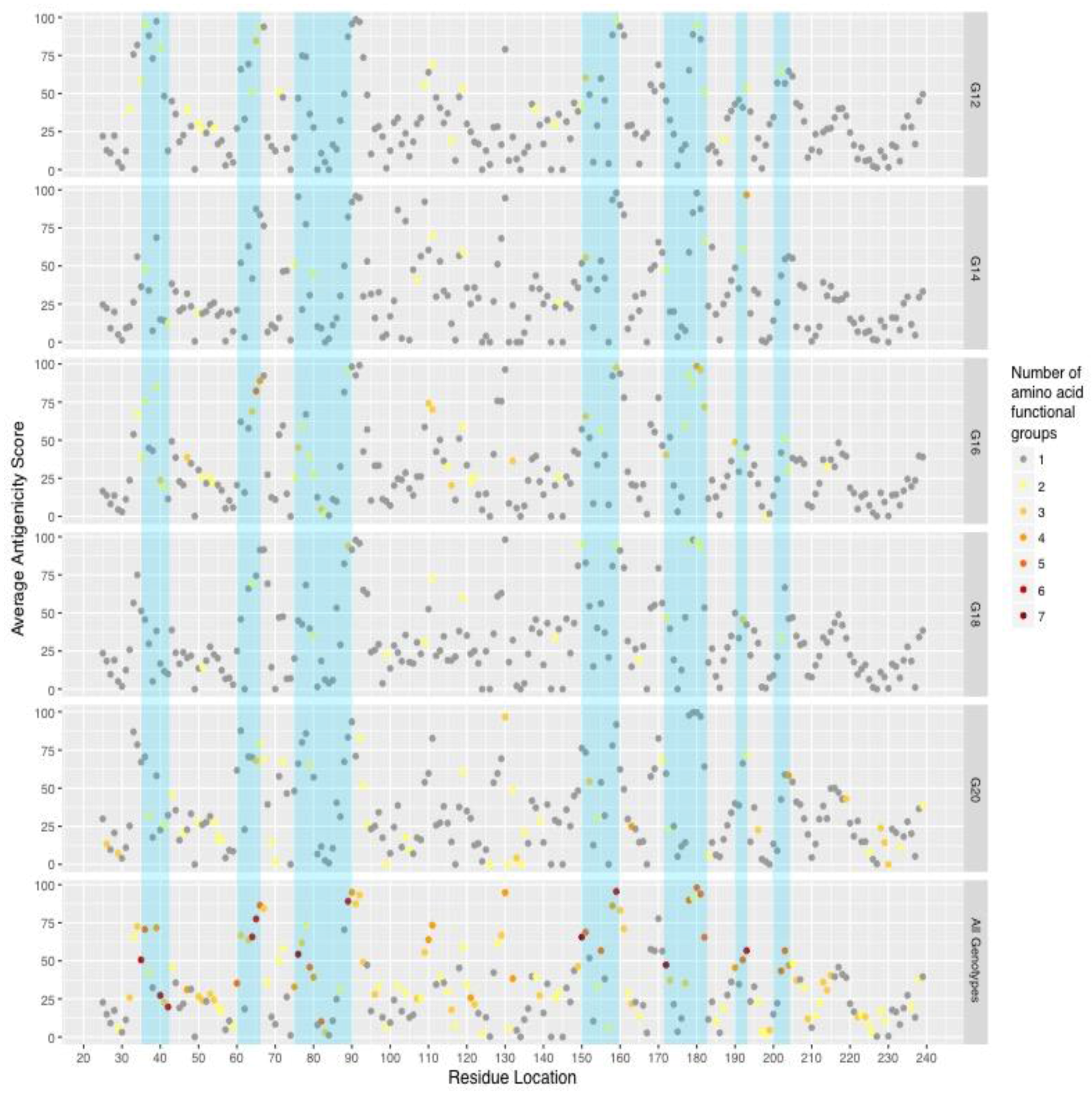
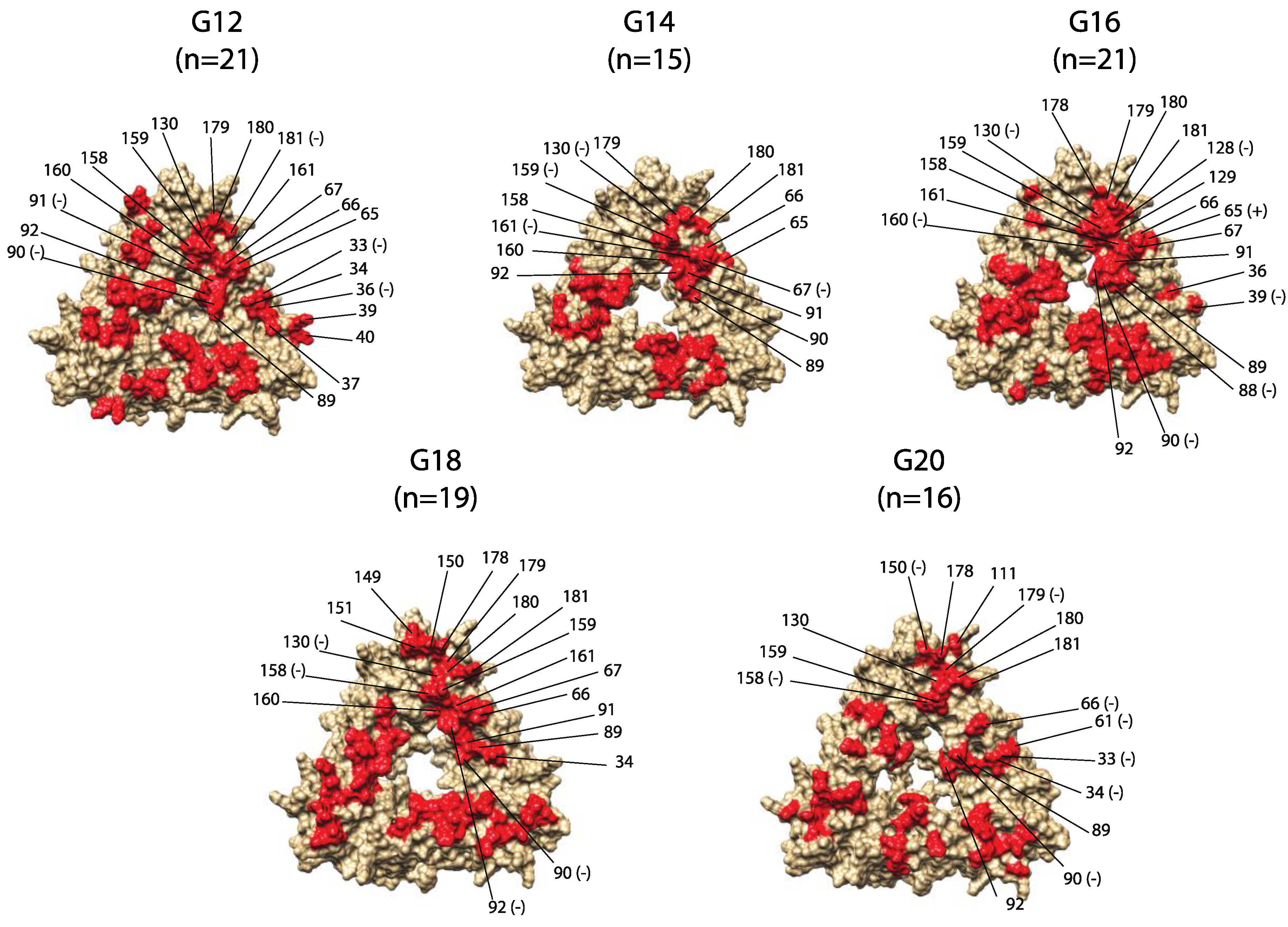
2. Results
3. Discussion
4. Materials and Methods
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Estes, M.K.; Greenberg, H.B. Rotaviruses. In Fields Virology; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1347–1401. ISBN 978-1-4698-7422-7. [Google Scholar]
- Bányai, K.; Kemenesi, G.; Budinski, I.; Földes, F.; Zana, B.; Marton, S.; Varga-Kugler, R.; Oldal, M.; Kurucz, K.; Jakab, F. Candidate new rotavirus species in Schreiber’s bats, Serbia. Infect. Genet. Evol. 2016, 48, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Matthijnssens, J.; Otto, P.H.; Ciarlet, M.; Desselberger, U.; Van Ranst, M.; Johne, R. VP6-sequence-based cutoff values as a criterion for rotavirus species demarcation. Arch. Virol. 2012, 157, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Mebus, C.A.; Underdahl, N.R.; Rhodes, M.B.; Twiehaus, M.J. Calf diarrhea (scours): Reproduced with a virus from field outbreak. Neb. Agric. Exp. Stn. Res. Bull. 1969, 233, 1–16. [Google Scholar]
- Mebus, C.A.; Underdahl, N.R.; Rhodes, M.B.; Twiehaus, M.J. Further studies on neonatal calf diarrhea virus. Proc. Annu. Meet. USA Anim. Health Assoc. 1969, 73, 97–99. [Google Scholar]
- Bridger, J.C.; Brown, J.F. Prevalence of antibody to typical and atypical rotaviruses in pigs. Vet. Rec. 1985, 116, 50. [Google Scholar] [CrossRef] [PubMed]
- Theil, K.W.; Saif, L.J.; Moorhead, P.D.; Whitmoyer, R.E. Porcine Rotavirus-Like Virus (Group B Rotavirus): Characterization and Pathogenicity for Gnotobiotic Pigs. J. Clin. Microbiol. 1985, 21, 340–345. [Google Scholar] [PubMed]
- Rodger, S.M.; Craven, J.A.; Williams, I. Demonstration of Reovirus-like Particles in Intestinal Contents of Piglets with Diarrhoea. Aust. Vet. J. 1975, 51, 536. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.M.; Pun, S.B.; Gauchan, P.; Yokoo, M.; Doan, Y.H.; Tran, T.N.H.; Nakagomi, T.; Nakagomi, O.; Pandey, B.D. The First Identification of Rotavirus B from Children and Adults with Acute Diarrhoea in Kathmandu, Nepal. Trop. Med. Health 2013, 41, 129–134. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tsunemitsu, H.; Morita, D.; Takaku, H.; Nishimori, T.; Imai, K.; Saif, L.J. First detection of bovine group B rotavirus in Japan and sequence of its VP7 gene. Arch. Virol. 1999, 144, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Theil, K.W.; Grooms, D.L.; McCloskey, C.M.; Redman, D.R. Group B rotavirus associated with an outbreak of neonatal lamb diarrhea. J. Vet. Diagn. Investig. 1995, 7, 148–150. [Google Scholar] [CrossRef] [PubMed]
- Munoz, M.; Alvarez, M.; Lanza, I.; Carmenes, P. Role of enteric pathogens in the aetiology of neonatal diarrhoea in lambs and goat kids in Spain. Epidemiol. Infect. 1996, 117, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Vonderfecht, S.L.; Lindsay, D.A.; Eiden, J.J. Detection of rat, porcine, and bovine group B rotavirus in fecal specimens by solid-phase enzyme immunoassay. J. Clin. Microbiol. 1994, 32, 1107–1108. [Google Scholar] [PubMed]
- Kuga, K.; Miyazaki, A.; Suzuki, T.; Takagi, M.; Hattori, N.; Katsuda, K.; Mase, M.; Sugiyama, M.; Tsunemitsu, H. Genetic diversity and classification of the outer capsid glycoprotein VP7 of porcine group B rotaviruses. Arch. Virol. 2009, 154, 1785. [Google Scholar] [CrossRef] [PubMed]
- Lahon, A.; Ingle, V.C.; Birade, H.S.; Raut, C.G.; Chitambar, S.D. Molecular characterization of group B rotavirus circulating in pigs from India: Identification of a strain bearing a novel VP7 genotype, G21. Vet. Microbiol. 2014, 174, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Marthaler, D.; Rossow, K.; Gramer, M.; Collins, J.; Goyal, S.; Tsunemitsu, H.; Kuga, K.; Suzuki, T.; Ciarlet, M.; Matthijnssens, J. Detection of substantial porcine group B rotavirus genetic diversity in the United States, resulting in a modified classification proposal for G genotypes. Virology 2012, 433, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Molinari, B.L. D.; Possatti, F.; Lorenzetti, E.; Alfieri, A.F.; Alfieri, A.A. Unusual outbreak of post-weaning porcine diarrhea caused by single and mixed infections of rotavirus groups A, B, C, and H. Vet. Microbiol. 2016, 193, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Otto, P.H.; Rosenhain, S.; Elschner, M.C.; Hotzel, H.; Machnowska, P.; Trojnar, E.; Hoffmann, K.; Johne, R. Detection of rotavirus species A, B and C in domestic mammalian animals with diarrhoea and genotyping of bovine species A rotavirus strains. Vet. Microbiol. 2015, 179, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Marthaler, D.; Homwong, N.; Rossow, K.; Culhane, M.; Goyal, S.; Collins, J.; Matthijnssens, J.; Ciarlet, M. Rapid detection and high occurrence of porcine rotavirus A, B, and C by RT-qPCR in diagnostic samples. J. Virol. Methods 2014, 209, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Bishop, R.F.; Tzipori, S.R.; Coulson, B.S.; Unicomb, L.E.; Albert, M.J.; Barnes, G.L. Heterologous protection against rotavirus-induced disease in gnotobiotic piglets. J. Clin. Microbiol. 1986, 24, 1023–1028. [Google Scholar] [PubMed]
- Gaul, S.K.; Simpson, T.F.; Woode, G.N.; Fulton, R.W. Antigenic relationships among some animal rotaviruses: Virus neutralization in vitro and cross-protection in piglets. J. Clin. Microbiol. 1982, 16, 495–503. [Google Scholar] [PubMed]
- Macmillan, L.; Ifere, G.O.; He, Q.; Igietseme, J.U.; Kellar, K.L.; Okenu, D.M.; Eko, F.O. A recombinant multivalent combination vaccine protects against Chlamydia and genital herpes. FEMS Immunol. Med. Microbiol. 2007, 49, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Banik, S.; Mansour, A.A.; Suresh, R.V.; Wykoff-Clary, S.; Malik, M.; McCormick, A.A.; Bakshi, C.S. Development of a Multivalent Subunit Vaccine against Tularemia Using Tobacco Mosaic Virus (TMV) Based Delivery System. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Sanekata, T.; Kuwamoto, Y.; Akamatsu, S.; Sakon, N.; Oseto, M.; Taniguchi, K.; Nakata, S.; Estes, M.K. Isolation of group B porcine rotavirus in cell culture. J. Clin. Microbiol. 1996, 34, 759–761. [Google Scholar] [PubMed]
- Wen, X.; Wei, X.; Ran, X.; Ni, H.; Cao, S.; Zhang, Y. Immunogenicity of porcine P[6], P[7]-specific ∆VP8* rotavirus subunit vaccines with a tetanus toxoid universal T cell epitope. Vaccine 2015, 33, 4533–4539. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Cao, D.; Jones, R.W.; Li, J.; Szu, S.; Hoshino, Y. Construction and characterization of human rotavirus recombinant VP8* subunit parenteral vaccine candidates. Vaccine 2012, 30, 6121–6126. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Pond, S.L. K. Detecting Individual Sites Subject to Episodic Diversifying Selection. PLOS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Zheng, D.; Zhang, C.; Zacharias, M. Prediction of antigenic epitopes on protein surfaces by consensus scoring. BMC Bioinform. 2009, 10, 302. [Google Scholar] [CrossRef] [PubMed]
- Vlasova, A.N.; Amimo, J.O.; Saif, L.J. Porcine Rotaviruses: Epidemiology, Immune Responses and Control Strategies. Viruses 2017, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Homwong, N.; Diaz, A.; Rossow, S.; Ciarlet, M.; Marthaler, D. Three-Level Mixed-Effects Logistic Regression Analysis Reveals Complex Epidemiology of Swine Rotaviruses in Diagnostic Samples from North America. PLoS ONE 2016, 11, e0154734. [Google Scholar] [CrossRef] [PubMed]
- Korva, M.; Knap, N.; Resman Rus, K.; Fajs, L.; Grubelnik, G.; Bremec, M.; Knapič, T.; Trilar, T.; Avšič Županc, T. Phylogeographic Diversity of Pathogenic and Non-Pathogenic Hantaviruses in Slovenia. Viruses 2013, 5, 3071–3087. [Google Scholar] [CrossRef] [PubMed]
- Lori, G.; Edel-Hermann, V.; Gautheron, N.; Alabouvette, C. Genetic Diversity of Pathogenic and Nonpathogenic Populations of Fusarium oxysporum Isolated from Carnation Fields in Argentina. Phytopathology 2004, 94, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Appel, D.J.; Gordon, T. Local and regional variation in populations of Fusarium oxysporum from agricultural field soils. Phytopathology 1994, 84, 786–791. [Google Scholar] [CrossRef]
- McDonald, S.M.; Matthijnssens, J.; McAllen, J.K.; Hine, E.; Overton, L.; Wang, S.; Lemey, P.; Zeller, M.; Van Ranst, M.; Spiro, D.J.; Patton, J.T. Evolutionary Dynamics of Human Rotaviruses: Balancing Reassortment with Preferred Genome Constellations. PLoS Pathog. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Dalkas, G.A.; Rooman, M. SEPIa, a knowledge-driven algorithm for predicting conformational B-cell epitopes from the amino acid sequence. BMC Bioinform. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Charles A Janeway, J.; Travers, P.; Walport, M.; Shlomchik, M.J. The Interaction of the Antibody Molecule with Specific Antigen; Garland Science: New York, NY, USA, 2001. [Google Scholar]
- Xia, Z.; Huynh, T.; Kang, S.; Zhou, R. Free-Energy Simulations Reveal that Both Hydrophobic and Polar Interactions Are Important for Influenza Hemagglutinin Antibody Binding. Biophys. J. 2012, 102, 1453–1461. [Google Scholar] [CrossRef] [PubMed]
- Doud, M.B.; Hensley, S.E.; Bloom, J.D. Complete mapping of viral escape from neutralizing antibodies. PLoS Pathog. 2017, 13, e1006271. [Google Scholar] [CrossRef] [PubMed]
- Lovelace, E.; Xu, H.; Blish, C.A.; Strong, R.; Overbaugh, J. The role of amino acid changes in the human immunodeficiency virus type 1 transmembrane domain in antibody binding and neutralization. Virology 2011, 421, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Naseer, O.; Jarvis, M.C.; Ciarlet, M.; Marthaler, D.G. Genotypic and epitope characteristics of group A porcine rotavirus strains circulating in Canada. Virology 2017, 507, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Zeller, M.; Patton, J.T.; Heylen, E.; De Coster, S.; Ciarlet, M.; Van Ranst, M.; Matthijnssens, J. Genetic Analyses Reveal Differences in the VP7 and VP4 Antigenic Epitopes between Human Rotaviruses Circulating in Belgium and Rotaviruses in Rotarix and RotaTeq. J. Clin. Microbiol. 2012, 50, 966–976. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.T.; Settembre, E.; Trask, S.D.; Greenberg, H.B.; Harrison, S.C.; Dormitzer, P.R. Structure of rotavirus outer-layer protein VP7 bound with a neutralizing Fab. Science 2009, 324, 1444–1447. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Qiu, J.; Zhu, Y.; Zhou, H.; Yu, L.; Ding, Y.; Zhang, L.; Guo, Z.; Dong, C. Molecular evolution of hepatitis B vaccine escape variants in China, during 2000–2016. Vaccine 2017, 35, 5808–5813. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.L.; Hughes, M.A. K. More Effective Purifying Selection on RNA Viruses than in DNA Viruses. Gene 2007, 404, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, C.A.; Méndez, E.; Zárate, S.; Isa, P.; López, S.; Arias, C.F. Integrin αvβ3 mediates rotavirus cell entry. Proc. Natl. Acad. Sci. USA 2000, 97, 14644–14649. [Google Scholar] [CrossRef] [PubMed]
- Zárate, S.; Espinosa, R.; Romero, P.; Guerrero, C.A.; Arias, C.F.; López, S. Integrin α2β1 Mediates the Cell Attachment of the Rotavirus Neuraminidase-Resistant Variant nar3. Virology 2000, 278, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Hewish, M.J.; Takada, Y.; Coulson, B.S. Integrins α2β1 and α4β1 Can Mediate SA11 Rotavirus Attachment and Entry into Cells. J. Virol. 2000, 74, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Graham, K.L.; Halasz, P.; Tan, Y.; Hewish, M.J.; Takada, Y.; Mackow, E.R.; Robinson, M.K.; Coulson, B.S. Integrin-Using Rotaviruses Bind 2 1 Integrin 2 I Domain via VP4 DGE Sequence and Recognize X 2 and V 3 by Using VP7 during Cell Entry. J. Virol. 2003, 77, 9969–9978. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y. Negative selection on neutralization epitopes of poliovirus surface proteins: Implications for prediction of candidate epitopes for immunization. Gene 2004, 328, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. circlize implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef] [PubMed]
- Pond, S.L.K.; Frost, S.D.W. Datamonkey: Rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 2005, 21, 2531–2533. [Google Scholar] [CrossRef] [PubMed]
- Pond, S.L.K.; Frost, S.D.W. Not So Different After All: A Comparison of Methods for Detecting Amino Acid Sites Under Selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A Fast, Unconstrained Bayesian AppRoximation for Inferring Selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- Conant, G.C.; Wagner, G.P.; Stadler, P.F. Modeling amino acid substitution patterns in orthologous and paralogous genes. Mol. Phylogenet. Evol. 2007, 42, 298–307. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | Year | |||||||
|---|---|---|---|---|---|---|---|---|
| 2011 | 2012 | 2013 | 2014 | 2015 | 2016 | 2017 | Total Sequences | |
| G8 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| G11 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 2 |
| G12 | 3 | 4 | 2 | 2 | 3 | 1 | 0 | 15 |
| G14 | 1 | 0 | 1 | 2 | 3 | 4 | 0 | 11 |
| G16 | 9 | 9 | 19 | 18 | 6 | 5 | 2 | 68 |
| G17 | 0 | 2 | 0 | 0 | 0 | 1 | 0 | 3 |
| G18 | 0 | 7 | 2 | 5 | 1 | 2 | 0 | 17 |
| G20 | 0 | 8 | 19 | 17 | 2 | 5 | 1 | 52 |
| G22 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 |
| G23 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 2 |
| G24 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 |
| G25 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| Total Sequences (Genotypes) | 13 (3) | 30 (5) | 46 (8) | 48 (8) | 16 (5) | 18 (6) | 3 (2) | 174 |
| Residue Location | G12 [n = 21] | G14 [n = 15] | G16 [n = 21] | G18 [n = 19] | G20 [n = 16] | Changed Properties | Conserved Properties |
|---|---|---|---|---|---|---|---|
| 33 | D − | D | D | D | N − | ||
| 34 | D | D | N | N | D/N − | ||
| 36 | T/N − | Q/T | N | N | N/T | Chemical composition | Volume, hydropathy |
| 37 | D | E | E | E | E | Iso-electric point | |
| 39 | K | K/R | T − | K | R/T/K | Chemical composition | |
| 40 | K/Q | E | Q/E | E | E | ||
| 61 | N | N | N | N | N − | ||
| 65 | N/E/D | E/D | Q/R+ | D | D/N | Volume | Polarity |
| 66 | N/D | N | N/S/D | N | N/K − | Polarity | |
| 67 | Y | Y − | Y | Y | Y | ||
| 76 * | H | Y − | T/I/V/M | Y | N/S | Iso-electric point | |
| 77 * | N | N | D/N | D | N/S | ||
| 78 * | Y | Y | Y | Y | F − | ||
| 88 * | V | V | V/I − | V | I | Hydropathy | |
| 89 | K | A | D | S/G/D | D/N | ||
| 90 | Y − | D | E − | D − | D − | Iso-electric point | |
| 91 | A − | P | P | P | P | Polarity | |
| 92 | Y | Y | W | F − | W | ||
| 102 * | E | E − | E | E | Q | ||
| 104 * | N | N | N | N | N | ||
| 109 * | A/T | V/A | A | A/T | A | ||
| 111 | G/R | K/N | N | E/D/N | E/G | ||
| 128 | S | S | S − | S | A | ||
| 129 | R/K | K | K/R | K/R | M/T | Polarity | Volume, iso-electric point, hydropathy |
| 130 | D | G − | G − | G − | D/N | ||
| 149 | L | L | L | T | T/I | Iso-electric point | |
| 150 | E/K | S | S | Y | P − | Polarity | Chemical composition |
| 151 | G/D | G/S/A/N | S/D/G | T | G/T/E/S | ||
| 158 | P | P | P | P − | P − | ||
| 159 | D/N | K − | E/K | E/D | N/K/D/S | Volume | |
| 160 | R | R | R − | R | R | ||
| 161 | R | R − | K | R | K/R | ||
| 170 * | F | F | F − | F − | F − | ||
| 178 | Y | Y | H | R | R | Polarity | |
| 179 | S | S | S | S | S − | ||
| 180 | N | D/E | T/A/K | S | Q/E | Iso-electric point | |
| 181 | N − | D | S/N | H/Y | N/S | ||
| 193 * | Q/P | L/D/N | P | P | P |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shepherd, F.K.; Murtaugh, M.P.; Chen, F.; Culhane, M.R.; Marthaler, D.G. Longitudinal Surveillance of Porcine Rotavirus B Strains from the United States and Canada and In Silico Identification of Antigenically Important Sites. Pathogens 2017, 6, 64. https://doi.org/10.3390/pathogens6040064
Shepherd FK, Murtaugh MP, Chen F, Culhane MR, Marthaler DG. Longitudinal Surveillance of Porcine Rotavirus B Strains from the United States and Canada and In Silico Identification of Antigenically Important Sites. Pathogens. 2017; 6(4):64. https://doi.org/10.3390/pathogens6040064
Chicago/Turabian StyleShepherd, Frances K., Michael P. Murtaugh, Fangzhou Chen, Marie R. Culhane, and Douglas G. Marthaler. 2017. "Longitudinal Surveillance of Porcine Rotavirus B Strains from the United States and Canada and In Silico Identification of Antigenically Important Sites" Pathogens 6, no. 4: 64. https://doi.org/10.3390/pathogens6040064
APA StyleShepherd, F. K., Murtaugh, M. P., Chen, F., Culhane, M. R., & Marthaler, D. G. (2017). Longitudinal Surveillance of Porcine Rotavirus B Strains from the United States and Canada and In Silico Identification of Antigenically Important Sites. Pathogens, 6(4), 64. https://doi.org/10.3390/pathogens6040064