ProtozoaDB 2.0: A Trypanosoma Brucei Case Study

Abstract
1. Introduction
2. Results
2.1. Protozoa Genomic Data
2.2. Proteome
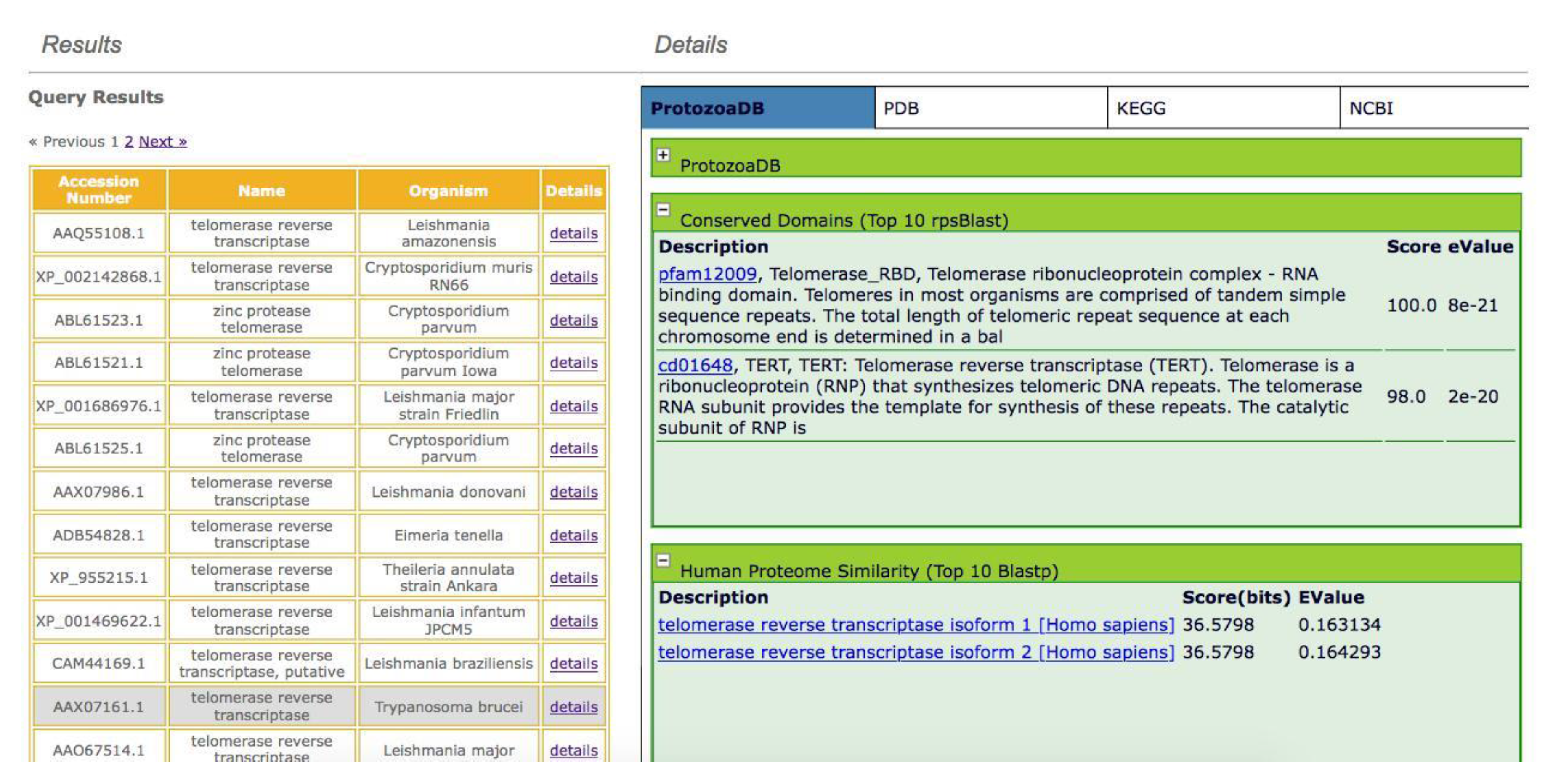
2.3. Homology
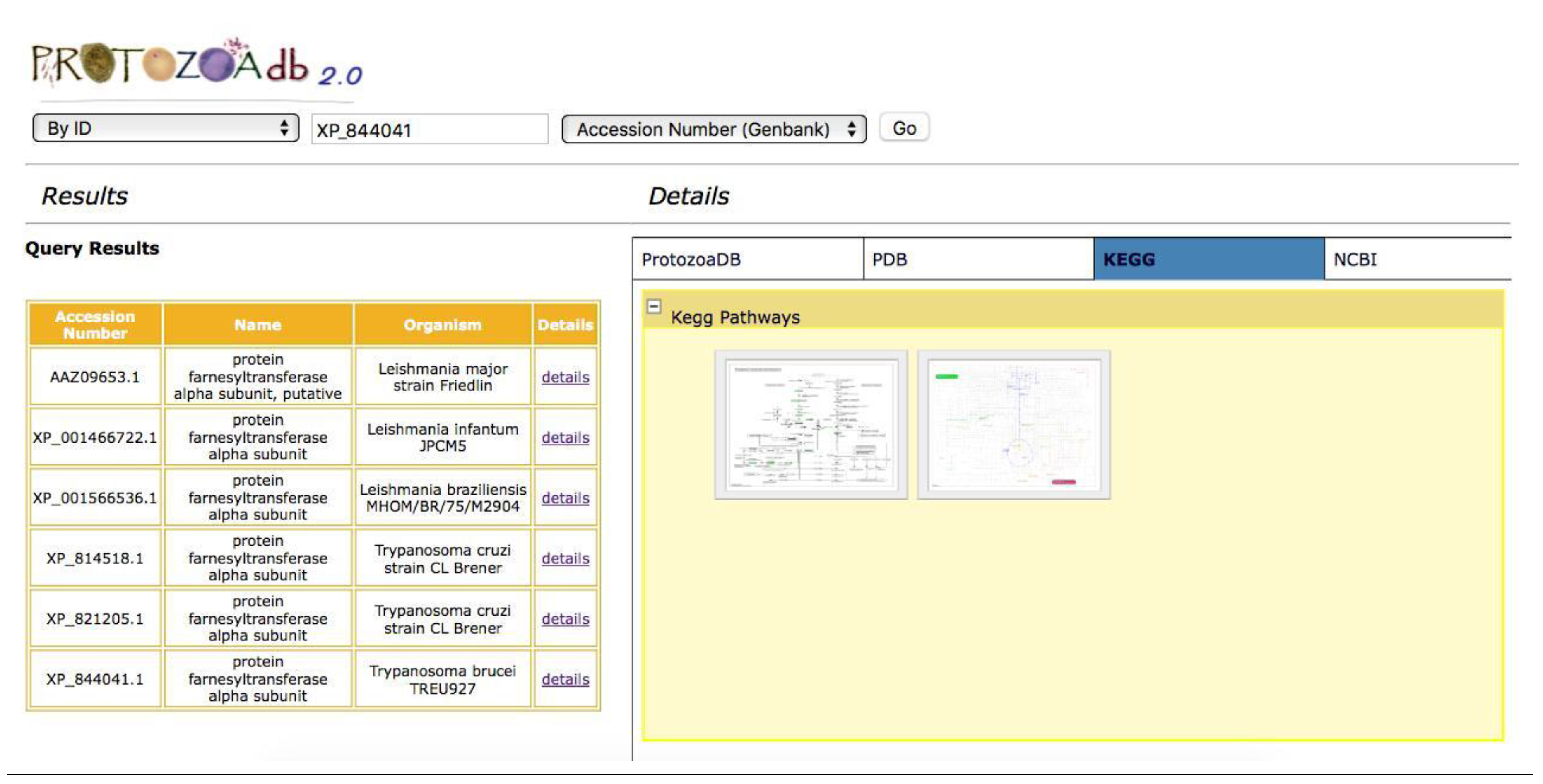
2.4. Metabolic Pathways
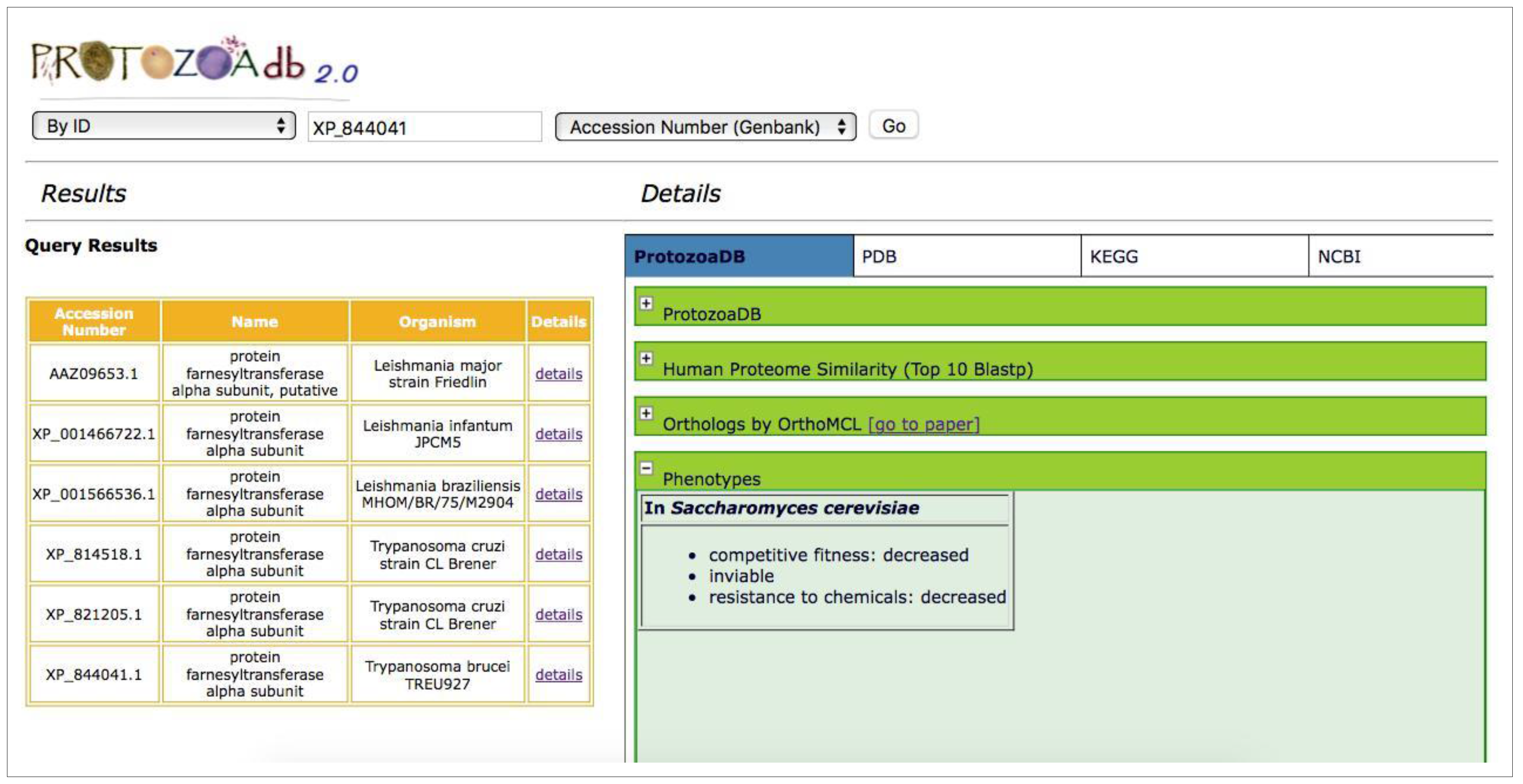
2.5. Phenotypes
2.6. How to Search
2.7. How to Search Using Our Web Service
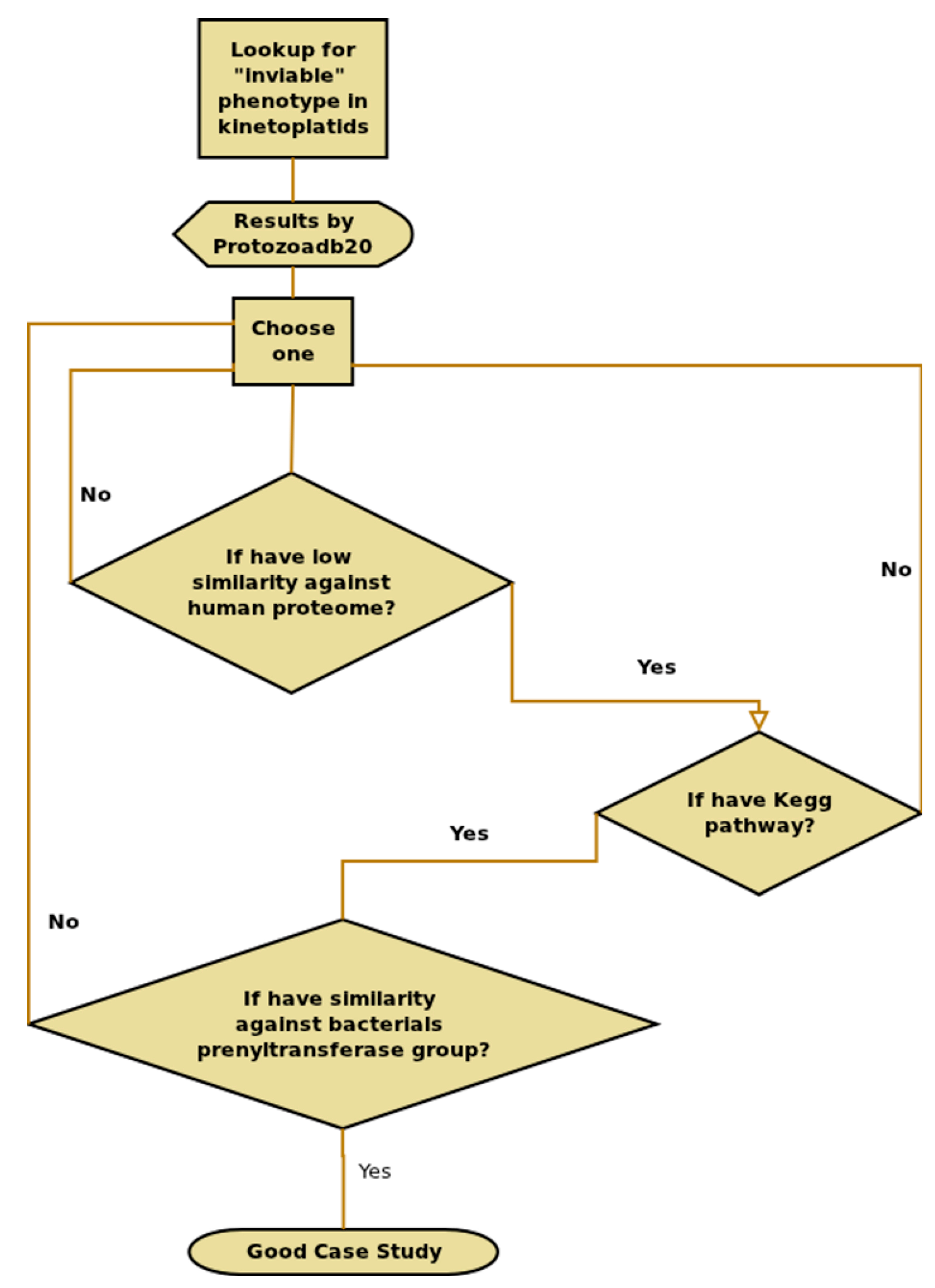
2.8. Information Extraction—T. brucei Case Study
2.9. Comparison with Another Information Extraction Tool
3. Discussion
3.1. T. Brucei Case Study
3.2. Comparison between ProtozoaDB 2.0 and EupathDB
4. Materials and Methods
4.1. Web Services
4.2. New Source Code
4.3. Data Acquisition
4.4. Preprocessing
4.5. Transformation
4.6. Analysis
- (1)
- Similarity against the human proteome: the system performs a query, through the web service; the human proteome is locally stored in the database, updated every six months, and returns the top ten hits, independent of the score or e-value.
- (2)
- Search for conserved domains: by running RPSBlast against the Conserved Domain Database (CDD) [39], the system returns the top ten results independent of the score or e-value.
- (3)
- Superfamily classification: the Superfamily database [14,40] has structural, functional, and evolutionary information of proteins from different genomes, including Protozoa. The new system performs a query through the web service, retrieving graphical information on the classification of superfamily.
- (4)
- (5)
- Metabolic pathways: the system performs a query for the KEGG Pathway database [10] to retrieve the metabolic pathways where a given protein participates, showing the maps and their interactions with other proteins participating in that pathway.
- (6)
- Literature: finally, the system performs two queries in Pubmed (http://www.ncbi.nlm.nih.gov/pubmed) to retrieve the original publication of the protein and other publications having relevance to the organism and the product.
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, D36–D42. [Google Scholar] [CrossRef] [PubMed]
- Brayton, K.A.; Lau, A.O.T.; Herndon, D.R.; Hannick, L.; Kappmeyer, L.S.; Berens, S.J.; Bidwell, S.L.; Brown, W.C.; Crabtree, J.; Fadrosh, D.; Feldblum, T.; Forberger, H.A.; et al. Genome Sequence of Babesia bovis and Comparative Analysis of Apicomplexan Hemoprotozoa. PLoS Pathog. 2007, 3, e148. [Google Scholar] [CrossRef] [PubMed]
- Heidel, A.J.; Lawal, H.M.; Felder, M.; Schilde, C.; Helps, N.R.; Tunggal, B.; Rivero, F.; John, U.; Schleicher, M.; Eichinger, L.; et al. Phylogeny-wide analysis of social amoeba genomes highlights ancient origins for complex intercellular communication. Genome Res. 2011, 21, 1882–1891. [Google Scholar] [CrossRef] [PubMed]
- Fritz-Laylin, L.K.; Prochnik, S.E.; Ginger, M.L.; Dacks, J.B.; Carpenter, M.L.; Field, M.C.; Kuo, A.; Paredez, A.; Chapman, J.; Pham, J.; et al. The genome of Naegleria gruberi illuminates early eukaryotic versatility. Cell 2010, 140, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Munõz, J.F.; Gallo, J.E.; Misas, E.; McEwen, J.G.; Clay, O.K. The eukaryotic genome, its reads, and the unfinished assembly. FEBS Lett. 2013, 587, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Kordjamshidi, P.; Roth, D.; Moens, M.F. Structured learning for spatial information extraction from biomedical text: Bacteria biotopes. BMC Bioinform. 2015, 16, 129. [Google Scholar] [CrossRef] [PubMed]
- Dávila, A.M.R.; Mendes, P.N.; Wagner, G.; Tschoeke, D.A.; Cuadrat, R.R.C.; Liberman, F.; Matos, L.; Satake, T.; Ocaña, K.A.C.S.; Triana, O.; et al. ProtozoaDB: Dynamic visualization and exploration of protozoan genomes. Nucleic Acids Res. 2008, 36, D547–D552. [Google Scholar]
- BioCreative, VI. Available online: http://www.biocreative.org (accessed on 16 July 2017).
- Krallinger, M.; Valencia, A.; Hirschman, L. Linking genes to literature: Text mining, information extraction, and retrieval applications for biology. Genome Biol. 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J.; Roos, D.S.; Christian, J.S., Jr. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Da Cruz, S.M.S.; Batista, V.; Silva, E.; Tosta, F.; Vilela, C.; Cuadrat, R.; Tschoeke, D.; Dávila, A.M.R.; Campos, M.L.M.; Mattoso, M. Detecting distant homologies on protozoans metabolic pathways using scientific workflows. Int. J. Data Min. Bioinform. 2010, 4, 256–280. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.; Pethica, R.; Zhou, Y.; Talbot, C.; Vogel, C.; Madera, M.; Chothia, C.; Gough, J. SUPERFAMILY–sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic Acids Res. 2009, 37, D380–D386. [Google Scholar] [CrossRef] [PubMed]
- Goto, N.; Prins, P.; Nakao, M.; Bonnal, R.; Aerts, J.; Katayama, T. BioRuby: Bioinformatics software for the Ruby programming language. Bioinform. Oxf. Engl. 2010, 26, 2617–2619. [Google Scholar] [CrossRef] [PubMed]
- Groth, P.; Weiss, B.; Pohlenz, H.D.; Leser, U. Mining phenotypes for gene function prediction. BMC Bioinform. 2008, 9, 136. [Google Scholar] [CrossRef] [PubMed]
- Groth, P.; Pavlova, N.; Kalev, I.; Tonov, S.; Georgiev, G.; Pohlenz, H.D.; Weiss, B. PhenomicDB: A new cross-species genotype/phenotype resource. Nucleic Acids Res. 2007, 35, D696–D699. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Pearson, W.R. Rapid and sensitive sequence comparison with {FASTP} and {FASTA}. Methods Enzymol. 1990, 183, 63–98. [Google Scholar] [PubMed]
- Marchler-Bauer, A.; Lu, S.; Anderson, J.B.; Chitsaz, F.; Derbyshire, M.K.; DeWeese-Scott, C.; Fong, J.H.; Geer, L.Y.; Geer, R.C.; Gonzales, N.R. CDD: A Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2011, 39, D225–D229. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.M.; Hong, E.L.; Amundsen, C.; Balakrishnan, R.; Binkley, G.; Chan, E.T.; Christie, K.R.; Costanzo, M.C.; Dwight, S.S.; Engel, S.R. Saccharomyces Genome Database: The genomics resource of budding yeast. Nucleic Acids Res. 2012, 40, D700–D705. [Google Scholar] [CrossRef] [PubMed]
- Maurer-Stroh, S.; Washietl, S.; Eisenhaber, F. Protein prenyltransferases. Genome Boil. 2003, 4, 212. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- European Bioinformatics Institute. Available online: http://www.ebi.ac.uk/about/terms-of-use (accessed on 16 July 2017).
- Aurrecoechea, C.; Barreto, A.; Basenko, E.Y.; Brestelli, J.; Brunk, B.P.; Cade, S.; Crouch, K.; Doherty, R.; Falke, D.; Fischer, S. EuPathDB: The eukaryoticpathogen genomics database resource. Nucleic Acids Res. 2017, 45, D581. [Google Scholar] [CrossRef] [PubMed]
- Anwar, T.; Gourinath, S. Deep Insight into the Phosphatomes of Parasitic Protozoa and a Web ResourceProtozPhosDB. PLoS ONE 2016, 11, e0167594. [Google Scholar] [CrossRef] [PubMed]
- Repchevsky, D.; Gelpi, J.L. BioSWR—Semantic Web Services Registry for Bioinformatics. PLoS ONE 2014, 9, e107889. [Google Scholar] [CrossRef] [PubMed]
- Velloso, H.; Vialle, R.A.; Ortega, J.M. BOWS (bioinformatics open web services) to centralize bioinformaticstools in web services. BMC Res. Notes 2015, 8, 206. [Google Scholar] [CrossRef] [PubMed]
- Papastergiou, A.; Tzekis, P.; Hatzigaidas, A.; Tryfon, G.; Ioannidis, D.; Zaharis, Z.; Kampitaki, D.; Lazaridis, P. A web-based melanoma image diagnosis support system using topic map and AJAX technologies. Inform. Health Soc. Care 2008, 33, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Yeung, D.; Boes, P.; Ho, M.W.; Li, Z. A Web application for the management of clinical workflow inimage-guided and adaptive proton therapy for prostate cancer treatments. J. Appl. Clin. Med. Phys. 2015, 16, 5503. [Google Scholar] [CrossRef] [PubMed]
- Ayong, L.; DaSilva, T.; Mauser, J.; Allen, C.M.; Chakrabarti, D. Evidence for prenylation-dependenttargeting of a Ykt6 SNARE in Plasmodium falciparum. Mol. Biochem. Parasitol. 2011, 175, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Pan, P.; Li, Y.; Li, D.; Yu, H.; Hou, T. Farnesyltransferase and geranylgeranyltransferase I:Structures, mechanism, inhibitors and molecular modeling. Drug Discov. Today 2015, 20, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Buckner, F.S.; Eastman, R.T.; Nepomuceno-Silva, J.L.; Speelmon, E.C.; Myler, P.J.; Van Voorhis, W.C.; Yokoyama, K. Cloning, heterologous expression, and substrate specificities of protein farnesyltransferasesfrom Trypanosoma cruzi and Leishmania major. Mol. Biochem. Parasitol. 2002, 122, 181–188. [Google Scholar] [CrossRef]
- Brunner, T.B.; Hahn, S.M.; Gupta, A.K.; Muschel, R.J.; McKenna, W.G.; Bernhard, E.J. Farnesyltransferase inhibitors: An overview of the results of preclinical and clinical investigations. Cancer Res. 2003, 63, 5656–5668. [Google Scholar] [PubMed]
- Shen, Y.; Qiang, S.; Ma, S. The Recent Development of Farnesyltransferase Inhibitors as Anticancer andAntimalarial Agents. Mini-Rev. Med. Chem. 2015, 15, 837–857. [Google Scholar] [CrossRef] [PubMed]
- Fielding, R.T. Architectural Styles and the Design of Network-Based Software Architectures. Ph.D Thesis, University of California, Irvine, CA, USA, 2000. [Google Scholar]
- Davidson, S.B.; Crabtree, J.; Brunk, B.; Schug, J.; Tannen, V.; Overton, C.; Stoeckert, C. K2/Kleisli and GUS: Experiments in Integrated Access to Genomic Data Sources. IBM Syst. J. 2001, 40, 512–531. [Google Scholar] [CrossRef]
- PostgreSQL. Available online: http://www.postgres.org (accessed on 16 July 2017).
- Marchler-Bauer, A.; Zheng, C.; Chitsaz, F.; Derbyshire, M.K.; Geer, L.Y.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Lanczycki, C.J. CDD: Conserved domains and protein three-dimensional structure. Nucleic Acids Res. 2013, 41, D348–D352. [Google Scholar] [CrossRef] [PubMed]
- De Lima Morais, D.A.; Fang, H.; Rackham, O.J.L.; Wilson, D.; Pethica, R.; Chothia, C.; Gough, J. SUPERFAMILY 1.75 including a domain-centric gene ontology method. Nucleic Acids Res. 2011, 39, D427–D434. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S. The Protein Data Bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Berman, H.M.; Kleywegt, G.J.; Nakamura, H.; Markley, J.L. The Protein Data Bank at 40: Reflecting on the past to prepare for the future. Struct. (Lond. Engl. 1993) 2012, 20, 391–396. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism/Strain |
|---|
| Babesia bovis T2Bo |
| Crypstosporidium parvum Iowa II |
| Cryptosporidium hominis TU502 |
| Cryptosporidium muris RN66 |
| Entamoeba dispar SAW760 |
| Entamoeba histolytica HM1:IMSS |
| Giardia lamblia ATCC 50803 |
| Leishmania braziliensis MHOM BR 75 M2904 |
| Leishmania infantum JPCM5 |
| Leishmania major Friedlin |
| Plasmodium berghei ANKA |
| Plasmodium chabaudi chabaudi AS |
| Plasmodium falciparum 3D7 |
| Plasmodium knowlesi strain H |
| Plasmodium vivax SaI 1 |
| Plasmodium yoelii yoelii 17XNL |
| Theileria annulata Ankara |
| Theileria parva Muguga |
| Toxoplasma gondii ME49 |
| Trichomonas vaginalis G3 |
| Trypanosoma brucei treu927 |
| Trypanosoma cruzi CL Brener |
| Functionalities | ProtozoaDB 2.0 | EuPathDB |
|---|---|---|
| Blast similarities against Homo sapiens | Available | n/a |
| Blast similarities against model organisms | Available | n/a |
| Blast similarities against protozoa species | Available | Available |
| Blast similarities against CDD | Available | n/a |
| Blast similarities against PDB | Available | n/a |
| Similarities against Intepro Domains | n/a | Available |
| KEGG metabolic pathways | Available | n/a |
| Protein structures by PDB | Available | n/a |
| Homology study: OrthoMCL | Available | Available |
| Homology study: KEGG orthologous | Available | n/a |
| Homology study: OrthoSearch | Available | n/a |
| Publications at PubMed | Available | n/a |
| Phenotype Search | Available | n/a |
| SNP Characteristics Search | n/a | Available |
| Genomic Position Search | n/a | Available |
| Information | URL |
|---|---|
| ProtozoaDB | http://services.biowebdb.org/howtouse |
| PDB | http://www.rcsb.org/pdb/software/rest.do |
| Kegg | http://www.kegg.jp/kegg/docs/keggapi.html |
| Pubmed (NCBI) | http://www.ncbi.nlm.nih.gov/books/NBK55693/ |
| Superfamily | http://supfam.cs.bris.ac.uk/SUPERFAMILY/web_services.html |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jardim, R.; Tschoeke, D.; Dávila, A.M.R. ProtozoaDB 2.0: A Trypanosoma Brucei Case Study. Pathogens 2017, 6, 32. https://doi.org/10.3390/pathogens6030032
Jardim R, Tschoeke D, Dávila AMR. ProtozoaDB 2.0: A Trypanosoma Brucei Case Study. Pathogens. 2017; 6(3):32. https://doi.org/10.3390/pathogens6030032
Chicago/Turabian StyleJardim, Rodrigo, Diogo Tschoeke, and Alberto M. R. Dávila. 2017. "ProtozoaDB 2.0: A Trypanosoma Brucei Case Study" Pathogens 6, no. 3: 32. https://doi.org/10.3390/pathogens6030032
APA StyleJardim, R., Tschoeke, D., & Dávila, A. M. R. (2017). ProtozoaDB 2.0: A Trypanosoma Brucei Case Study. Pathogens, 6(3), 32. https://doi.org/10.3390/pathogens6030032