Abstract
African trypanosomes cause devastating disease in sub-Saharan Africa in humans and livestock. The parasite lives extracellularly within the bloodstream of mammalian hosts and is transmitted by blood-feeding tsetse flies. In the blood, trypanosomes exhibit two developmental forms: the slender form and the stumpy form. The slender form proliferates in the bloodstream, establishes the parasite numbers and avoids host immunity through antigenic variation. The stumpy form, in contrast, is non-proliferative and is adapted for transmission. Here, we overview the features of slender and stumpy form parasites in terms of their cytological and molecular characteristics and discuss how these contribute to their distinct biological functions. Thereafter, we describe the technical developments that have enabled recent discoveries that uncover how the slender to stumpy transition is enacted in molecular terms. Finally, we highlight new understanding of how control of the balance between slender and stumpy form parasites interfaces with other components of the infection dynamic of trypanosomes in their mammalian hosts. This interplay between the host environment and the parasite’s developmental biology may expose new vulnerabilities to therapeutic attack or reveal where drug control may be thwarted by the biological complexity of the parasite’s lifestyle.
Keywords:
Trypanosoma brucei; transmission; stumpy form; differentiation; life-cycle; quorum sensing 1. Introduction
Persistence in regularly changing environments demands that vector-transmitted pathogens rapidly adapt to fluctuating conditions. Trypanosoma brucei, similar to many eukaryotic parasites, encounters different environments as it transitions between its mammalian host and the tsetse fly vector, Glossina spp. In these different environments, distinct T. brucei life cycle forms, specialised for the various niches encountered, have evolved to optimise parasite survival and to ensure transmission to the next host (Figure 1).
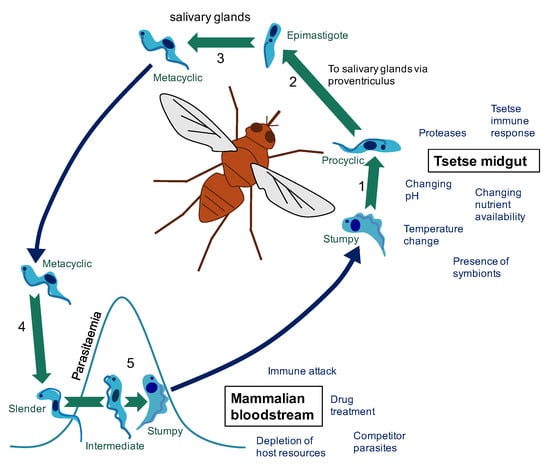
Figure 1.
The changing environments of the T. brucei life cycle. (1) Following a tsetse fly blood meal on an infected mammal (blue arrow), T. brucei stumpy cells reaching the tsetse midgut differentiate to procyclic cells. (2) Development of T. brucei proceeds through the tsetse midgut to the proventriculus. (3) Differentiation to epimastigote cells is followed by production of mammalian-infective metacyclic cells in the salivary gland. (4) On transfer to a new mammalian host via a tsetse fly bite (blue arrow), metacyclic cells differentiate to bloodstream forms. (5) In the bloodstream, proliferative slender cells elevate the parasitaemia until accumulation of a density-dependent signal triggers differentiation to the cell-cycle arrested stumpy form (via an intermediate stage). The stumpy form of T. brucei faces a particular challenge in that it must survive long enough in the bloodstream to be transmitted, but it must also survive long enough in the tsetse midgut to differentiate to the next life cycle stage and establish infection. These different environments pose different challenges that must be overcome to enable the continuation of the parasite life cycle. For example, in the mammalian host (red section), parasites must evade immune attack to ensure parasite survival and avoid excessive exploitation of host resources to ensure the host survives long enough to enable successful transmission to the tsetse fly vector. On uptake to the tsetse fly midgut (orange section), the parasites face a shift in nutrient availability and temperature among other challenges. For a review of factors effecting establishment of tsetse fly midgut infection see [1].
The challenges presented by the distinct environments encountered during the life cycle differ. In the mammalian bloodstream, for example, the parasite must face a host immune response that is constantly trying to control the infection. To evade this control and to thrive extracellularly within the mammalian host bloodstream, trypanosomes have evolved the extraordinary capacity to overcome a barrage of host innate and adaptive immunological defences via the expression, and switching, of their expressed Variant Surface Glycoprotein (VSG) coat, a process termed antigenic variation [2]. The periodic destruction of antigenic variants, followed by the outgrowth of antigenically distinct variants, contributes to the undulating waves of parasitaemia typical of trypanosome infection.
Within each of these cyclical waves, a density-dependent developmental switch represents another important evolutionary adaptation, both regulating parasite virulence and pre-adapting the parasite for uptake by the tsetse fly. Infections are maintained in the host bloodstream by rapidly proliferating morphologically “slender” cells. Allowed to replicate unchecked, slender parasites would rapidly kill the host [3]. Consequently, it is important that parasites regulate their growth in vivo to promote longevity of the host and maximize their transmission potential [4,5]. Such regulation is achieved by a developmental switch initiated upon reaching a critical parasite density. Specifically, proliferative slender cells respond to the accumulation of a small soluble parasite-derived factor, stumpy induction factor (or factors) (SIF), and differentiate to specialized cell-cycle arrested “stumpy” cells [6,7]. As growth-arrested stumpy cells accumulate during the ascending parasitaemia, the parasite population growth is restricted thereby prolonging the infection and increasing the opportunity for transmission [6,7,8]. Parasite strains that respond to this density-mediated growth control by differentiating to stumpy cells are described as pleomorphic. Extensive passage of laboratory strains can, however, select against differentiation to stumpy cells, resulting in “monomorphic” strains that do not arrest as transmissible forms in response to parasite density [9].
Beyond its cell-cycle arrest, the stumpy form of T. brucei has characteristics that distinguish it from the slender form, fulfilling its specialised role in transmission to the insect vector. In this review, we begin by describing the developments in our understanding of the nature of the different forms of T. brucei in the mammalian host, from initial morphological descriptions to detailed molecular characterisation. Secondly, we discuss investigations into the mechanism coordinating differentiation from slender to stumpy cells, considering how historical findings have provided tools to further elucidate this process. Finally, we reflect on outstanding questions relating to T. brucei differentiation and infection dynamics in the mammalian host.
2. Characterisation of Slender and Stumpy Cells
Differences in the morphology of Trypanosoma brucei spp. in the mammalian bloodstream were recognised in the early 1900s with descriptions and drawings of slender, intermediate and stumpy cells by Bruce [10]. These descriptions were followed by the observation that the stumpy form was apparently the morphotype capable of establishing infection of the tsetse fly vector [11,12]. Subsequent research sought to answer why the stumpy form was able to survive in the tsetse midgut and thus continue the parasite’s life cycle.
On uptake by the vector, parasites enter a challenging environment in the tsetse midgut. Slender cells are killed rapidly and it is stumpy cells that survive to differentiate to the next life cycle stage [13]. Characteristics of stumpy cells that likely contribute to their survival in the fly midgut include their relative resistance to acidic and proteolytic stress [14]. For example, the differential response to mild acid conditions (pH 5.5) has been used as a selection to monitor the differentiation to stumpy cells in some studies [15], as slender cells are sensitive and stumpy cells are resistant. Moreover, protease treatment has been shown to act as a trigger for bloodstream forms to differentiate to procyclic cells [16,17,18]. Likewise, pretreatment in mild-acid conditions (pH 5.5) has been shown to stimulate stumpy cells to differentiate to procyclic cells at 27 °C [19].
Different host environments also provide alternative nutritional opportunities for a parasite. For instance, in the mammalian bloodstream T. brucei can exploit an abundant glucose supply. On uptake by the tsetse fly, however, proline replaces glucose as the most readily available energy source [20], and an intact proline metabolic pathway has been shown to be essential for successful T. brucei colonisation of the tsetse midgut [21]. Correspondingly, procyclic cells inhabiting the tsetse midgut possess a more elaborate and active mitochondrion than slender bloodstream forms [22]. In preparation for this, differentiation in the bloodstream from the slender form to the stumpy form is accompanied by activation of the mitochondrion, as evidenced by detection of NAD diaphorase activity and the ability of intermediate and stumpy cells to use the Krebs cycle intermediate α-keto-glutaric acid (α-KGA) to maintain motility in the absence of glucose [23]. NADH dehydrogenase (diaphorase) activity has remained an accepted (though qualitative and somewhat ambiguous) marker for identifying stumpy bloodstream cells, but has more recently been superseded by the use of mitochondrial dyes such as MitoTracker® (Molecular probesTM) [24]. A further intracellular rearrangement occurring during the differentiation to stumpy cells is the repositioning of the lysosome. In slender cells a single lysosome is positioned equidistant between the nucleus and kinetoplast at the posterior end of the cell, whereas differentiation to stumpy cells is accompanied by movement of the lysosome to the anterior side of the nucleus [25]. An additional remodelling of the endocytic system that occurs during differentiation is the down-regulation of the haptoglobin-haemoglobin receptor in stumpy cells [25].
Once stumpy cells have been ingested by the tsetse fly they must be ready to perceive the signals that trigger differentiation to the proliferative procyclic form adapted for the colonisation of the midgut. Cold shock (i.e., a reduction from 37 °C to 20 °C) is able to sensitise stumpy (but not slender) parasites for perception of the differentiation signals cis-aconitate and citrate [26]. A carboxylate transporter protein family, known as Proteins Associated with Differentiation (PAD), is required for the response to this citrate cis-aconitate differentiation signal [27]. Thus, PAD2 expression is up-regulated at 20 °C and the protein is redistributed from the flagellar pocket to the parasite surface, enabling the cold-stimulated perception of the differentiation signal. PAD1 protein, in contrast, was found to be expressed specifically on the surface of stumpy, but not slender, bloodstream cells and is now used extensively in differentiation studies as a molecular marker for stumpy parasites. For example, in a study of chronic T. brucei infections in mice, intermediate and stumpy cells were found to dominate later stages of infection [8], the relative expression of PAD1 mRNA detected by qRT-PCR being used as a tool to reflect the transmission capacity of the population. PAD1 mRNA expression is detectable in the intermediate cells that precede morphological transformation to stumpy cells, this being followed by protein expression as mature stumpy cells arise, the regulatory signals governing progressive activation having been localised in the PAD1 3′UTR [28].
Another group of proteins with strong developmental regulation are the Expression Site Associated Gene (ESAG) 9 family [29] which are specifically expressed in stumpy, but not slender cells. Although originally identified as components of some VSG expression sites [30], ESAG9 genes are rare expression site (ES) components [31], being frequently found in non-expression site locations. The function fulfilled by this family of proteins remains unclear, but they appear to be secreted by the parasite and could therefore interact with the mammalian immune system or assist stumpy form transmission upon entry into the fly. Regions in the 3′UTRs of ESAG9 [32] and PAD1 [28] with involvement in the developmental regulation of these stumpy-expressed proteins have been identified. Moreover, a cell line engineered to place expression of a reporter gene under the control of the PAD1 3′UTR has been developed as a useful tool to study differentiation from slender to stumpy cells. This cell line was successfully used in a high-throughput screen to identify a compound able to induce partial differentiation to stumpy cells [33], whereas a PAD1 3′UTR linked GFP reporter has also been used in cytological studies of stumpy cells in adipose tissue [34]. Recently, knowledge of the stumpy-specific regulation of ESAG9 was successfully applied to identify a protein that negatively regulates the expression of stumpy-specific genes [35]. This utilised a screen exploiting a cell line containing both a tetracycline-inducible genome-wide RNAi library [36] and a drug resistance gene under the control of the 3′UTR of ESAG9. The screen identified REG9.1, a molecule with a predicted RNA binding motif, whose silencing by RNAi resulted in derepression of many ESAG9 genes as well as the mRNAs for two novel predicted cell-surface families (Family 5 and Family 7) that were also found to be enriched in stumpy cells [35].
The most obvious distinction between slender and stumpy cells of T. brucei is that a slender population is proliferative, whereas stumpy cells are arrested in G0/G1 of their cell cycle [37]. In addition to extending host survival, the cell cycle arrest displayed by stumpy cells contributes to their competence for transmission. For instance, it was proposed that there was a point in the cell cycle (G0/G1) when cells were receptive to the signal to differentiate to procyclic cells after which they re-enter the cell cycle [38]. This co-ordination of cell cycle position and differentiation capacity ensures that the organellar repositioning events that accompany differentiation can be achieved without perturbation of the precisely orchestrated cytological events that accompany the parasite’s cell division [39,40]. It also explains why uniform populations of stumpy cells already arrested at this point of the cell cycle are able to differentiate synchronously, unlike asynchronous monomorphic bloodstream forms [41].
The efficiency with which stumpy cells differentiate synchronously to procyclic cells in vitro provides a useful and physiologically relevant assay to monitor the abundance of stumpy cells in a bloodstream parasite population. This assay can be effectively performed in vitro using cis-aconitate, combined with a reduction in temperature from 37 °C to 27 °C, to stimulate differentiation [41,42,43]. The changes accompanying differentiation to procyclic cells include replacement of the VSG coat with a procyclin surface coat [44,45,46], repositioning of the mitochondrial genome (the kinetoplast) [40] and cell cycle re-entry [38,47]. Thus, the tools exist to effectively test the capacity of a population of bloodstream trypanosomes to synchronously differentiate to procyclic cells, helping to distinguish physiologically relevant stumpy cells from (for example) RNAi phenotypes that perturb cell shape or proliferation and generate cells superficially similar to stumpy morphology.
Differentiation from stumpy to procyclic cells triggered by citrate/cis-aconitate or mild acid treatment is signalled via a protein phosphorylation cascade [18]. TbPTP1, a protein tyrosine phosphatase [48], dephosphorylates and inactivates the DxDxT class serine-threonine phosphatase TbPIP39 (TbPTP1-interacting protein, 39kDa) [49] to prevent differentiation of stumpy cells in the bloodstream. Once parasites reach the appropriate environment of the tsetse midgut, they perceive the differentiation signal, TbPTP1 is inactivated and TbPIP39 is phosphorylated (and so activated) and translocated to the glycosome. TbPIP39 mRNA is expressed at highest levels in stumpy cells. However, while barely detectable in slender cells, TbPIP39 protein is detected in stumpy cells but protein expression is highest in procyclic cells [49]. The expression of proteins, such as TbPIP39, required for differentiation to the next life cycle stage is another feature of stumpy cells that prepares them for transmission. Indeed, the detection of increased TbPIP39 transcript abundance has been used as an indicator of the presence of stumpy cells in a bloodstream population of T. b. rhodesiense [50], and this can supplement the use of other stumpy markers such as PAD1 expression. Likewise, the protein kinase NRKA/B is a key regulator of differentiation when parasites enter the tsetse fly and this molecule is exclusively expressed in stumpy cells, providing another means to identify this cell type [51,52].
Further differences between slender and stumpy bloodstream cells have been described in how these cells interact with the host immune system. Slender cells of T. brucei can switch their expressed VSG to one that is antigenically distinct and outgrow as new antigenic variants. This enables survival of a subset of switched cells when the host develops an immune response against the dominant VSG expressed by the population ([22], and reviewed in [53]). Stumpy cells, in contrast, do not switch to express novel VSG [54] and—being irreversibly arrested—cannot establish new variant populations. However, stumpy cells have a higher rate of endocytosis than slender cells and are better able to rapidly clear IgG-VSG from the cell surface to delay complement-mediated lysis [55,56]. This may enable stumpy cells to survive for long enough to increase their probability of transmission.
T. brucei VSG expression is under strict control with transcription active from only one expression site at a time, maintaining monoallelic VSG expression [31,57]. A link between antigenic variation and life cycle progression was proposed when it was found that overexpression of an exogenous VSG in monomorphic T. brucei could stimulate an accumulation of the cell population in G1 of the cell cycle, expression of PAD1 and an enhanced ability to differentiate to procyclic cells in response to cis-aconitate treatment [58]. This effect was mediated through repression of the active ES, and linked to the concomitant silencing of ESAGs 1, 2 and 8 since RNAi targeting these transcripts reproduced the phenotype. However, this effect must be distinct from the quorum-sensing mediated differentiation from slender to stumpy cells since the phenotype caused by overexpression of the exogenous VSG was reversible. This contrasts with density-induced slender to stumpy differentiation which is irreversible.
Encouraged by the findings in monomorphic cells, Zimmermann et al. (2017) recently tested the hypothesis that transcriptional attenuation at the ES could elicit life cycle progression in biologically-relevant pleomorphic cells in vitro [59]. Here, ectopic overexpression of a second VSG generated two populations of cells with distinct growth phenotypes, either those which rapidly ceased proliferation upon induction or those that continued to divide. The growth-arrested cells exhibited complete transcriptional attenuation of the ES, whilst those that continued to divide did not. Moreover, VSG overexpression-induced ES attenuation caused cells to accumulate in G1 and upregulate the expression of stumpy-specific proteins, such as PAD1, in a density (and therefore SIF) independent manner. Upon exposure to cis-aconitate, cells could synchronously differentiate to procyclic cells, and were capable of establishing mature tsetse fly infections demonstrating their physiological relevance. Cells which remained proliferative upon overexpression of a second VSG, however, remained slender and, though the initial VSG was silenced, the ES remained in an incompletely attenuated state. The authors proposed that, upon switching to a new ES with defective function, the parasite may respond in one of two ways: if the activity of the initial ES remains above a critical threshold, this state is maintained and the parasite continues to proliferate; if, however, the ES-activity has fallen below this threshold, the parasite differentiates to the growth-arrested stumpy form in a density-independent manner. This mechanism was proposed to provide a potential escape for parasites that switch to an ES insufficient to fulfil requirements in a given host environment, although the physiological relevance of this for overall parasite transmission remains to be established.
Advances in technology have allowed global analyses of differences between life cycle forms in terms of expressed transcripts, protein abundance and phosphorylation status. In the stumpy form, reduced protein synthesis, possibly regulated through translation initiation [60], accompanies cell cycle arrest [61]. Accordingly, transcripts such as histones associated with an active cell cycle, as well as glycolytic pathway components and cytoskeletal transcripts were found to be down-regulated in stumpy cells [62,63,64]. On the other hand, transcripts involved in protein synthesis were observed to be up-regulated in procyclic cells [62]. Transcripts escaping the generalised translational repression in stumpy cells were identified from polysomal material [61]. Thus, components of oxidative phosphorylation and amino acid metabolism, amino acid transporters and mitochondrial transcripts were enriched in stumpy cells, matching expectation for the life cycle stage primed for transmission to the insect vector.
In T. brucei transcript levels do not necessarily correspond to protein levels, as much regulation of gene expression occurs post-transcriptionally through binding of regulators to (predominantly) the 3′UTR of a particular gene, with consequences for transcript stability and translational efficiency [65]. Proteomic analyses are therefore required to compare the protein composition of various T. brucei life cycle forms. RNA-binding proteins, cell surface proteins and proteins involved in mitochondrial and glycosomal energy metabolism have been found to vary in abundance between cultured bloodstream and procyclic form T. brucei [66,67]. Such studies often used SILAC (stable isotope labelling by amino acids in culture) and this methodology has been extended to analysis of protein abundance in slender, stumpy and procyclic cells of the pleomorphic cell line T. brucei AnTat 1.1 [68]. Here, it was observed that mitochondrial proteins were more abundant in stumpy and procyclic cells than in slender cells, with many protein abundance changes already being evident between the slender and stumpy stages. It is important to note that the increase in abundance of mitochondrial proteins in procyclic cells observed in this study was of a greater magnitude than in a previous study [67] that did not use a pleomorphic strain of T. brucei. This emphasises the need to use pleomorphic strains, competent to complete the life cycle, to fully characterise the features of different life cycle forms. Further insight into the timings of protein abundance changes during the T. brucei life cycle has been obtained by following changes in protein abundance and phosphorylation before and after the point of commitment to differentiation from bloodstream stumpy cells to procyclic cells [51]. Just 3 h following stimulation of differentiation with cis-aconitate, proteins associated with the procyclic form cell surface and developmental changes in metabolism were upregulated. Additionally, a change in the phosphorylation status of eukaryotic initiation factors was detected consistent with reinitiation of protein synthesis early in differentiation [62]. Likewise, also using the synchronous differentiation of stumpy cells, Dejung et al. found proteins indicative of a changing cell surface and metabolism increased in abundance 2 h following induction of differentiation [69]. Moreover, some proteins enriched in procyclic cells, such as PAD2 and TbPIP39 were already more abundant in stumpy cells relative to slender cells.
As a consequence of the extensive differences in the characteristics and developmental potential of slender and stumpy cells of Trypanosoma brucei it is important to be able to identify these forms within a population of trypanosomes under study (Figure 2). For instance, in cases where variations in gene expression over time or in different conditions are being reported in bloodstream parasites it is essential to consider whether perceived differences may be a result of varying proportions of differentiated cells, which may or may not be a consequence of the conditions under test. For example, in the recent description of transcripts in T. brucei that show circadian control [70], a reporter cell line with GFP expression under the control of the PAD1 3′UTR was used to confirm that the majority of the trypanosome cultures under study were slender (GFP-negative). This confirmed that variations in the transcript abundances observed were not contributed by variations in the proportion of stumpy cells.
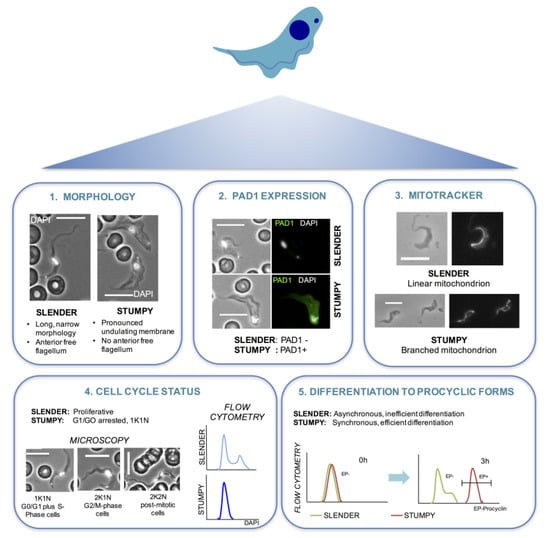
Figure 2.
Characteristics of a stumpy cell. A number of assays can be used to test the proportion of cells in a T. brucei bloodstream form population that are slender or stumpy. (1) Firstly, stumpy cells are clearly morphologically distinct from slender cells. The representative cells shown have been labelled with the DNA stain DAPI (white) to highlight the position of the nucleus and kinetoplast in each case. (2) Secondly, stumpy cells express the PAD1 protein on their surface [27]. PAD1 positive cells can be identified using the PAD1 antibody by immunofluorescence microscopy or Western blotting. PAD-based reporter assays have also been established for enzymatic or cytological quantitation. (3) Stumpy cells also have a more elaborated mitochondrion than slender cells [23] and this can be detected by incubating the cells with mitotracker dye [24] before fixing and visualising the cells with a fluorescence microscope. (4) During the cell cycle, T. brucei cells first segregate their replicated mitochondrial genome (kinetoplast, K) and then their nucleus (N) prior to cytokinesis to generate two daughter cells [71]. Stumpy cells are arrested in G1/G0 of the cell cycle and have 1 kinetoplast and 1 nucleus (1K1N) [37]. Thus, differentiation to a stumpy population is accompanied by an accumulation of 1K1N cells, and this can be detected by staining with an appropriate DNA dye (e.g., DAPI) and KN scoring by immunofluorescence microscopy or flow cytometry (a completely stumpy population will have a single dominant peak corresponding to G1/G0). (5) Finally, stumpy cells, unlike slender cells, are able to differentiate synchronously to procyclic cells in culture [41], when differentiation is triggered by incubating cells at 27 °C in SDM-79 medium containing 6 mM cis-aconitate. Differentiation to procyclic cells is indicated by expression of EP procyclin [72] and can be detected using an EP procyclin antibody and flow cytometry. Maximal expression of EP procyclin is detectable 3 h after exposure of stumpy cells to cis-aconitate [51].
3. Tools to Understand Life Cycle Development
In parallel to the extensive characterisation of slender and stumpy form parasites, there has also been significant progress in understanding of the mechanisms regulating differentiation from slender to stumpy cells. This has been assisted by tools and technologies that enable dissection of the underlying processes.
Early differentiation work focused on the anti-proliferative properties of the repurposed anti-cancer drug DL-α-difluoromethylornithine (DFMO), a specific irreversible inhibitor of the enzyme responsible for catalysing the first reaction in the polyamine biosynthesis pathway, ornithine decarboxylase (ODC) [73,74]. Exposure of mammalian cells to DFMO causes a rapid depletion of intracellular levels of the polyamines spermidine and putrescine resulting in the cessation of cellular proliferation, though largely in the absence of cell death [73,75]. Furthermore, many studies have attested to the importance of the role that polyamines, in particular spermidine and putrescine, play in the stimulation and regulation of mammalian cell differentiation (reviewed in [76]). Based on these observations, early studies sought to explore the possibility that polyamine depletion upon DFMO treatment could function to regulate T. brucei slender to stumpy differentiation, and, moreover whether DFMO induced differentiation events, and the subsequent morphological and physiological changes, were relevant to those seen in natural pleomorphic populations [77]. After treatment with DFMO, monomorphic T. brucei strains, grown either in vitro or in vivo, quickly became deficient in polyamines and stopped proliferating with no apparent cytotoxic effect [77,78,79,80]. In addition, monomorphic slender cells transformed morphologically to a smaller rounder cell form with a shortened flagellum, suggestive of a stumpy-like form [77]. While Bacchi et al. [78] were unable to isolate true stumpy cells from DFMO treated animals, possibly due to clearance by the immune system, de Gee et al. [79] demonstrated enhanced activity of NAD diaphorase in monomorphic cells isolated from infections following DFMO treatment, suggesting activation of the mitochondrion in preparation for transmission to the tsetse fly. Giffin et al. [77] reported similar morphological and physiological changes in response to DFMO when culturing monomorphic strains in vitro. However, though the DFMO-treated cells appeared to have increased mitochondrial activity and some stumpy characteristics, they were not able to efficiently transform to procyclic cells in SDM79 medium [77,80]. Moreover, treatment with an exogenous source of putrescine was shown to relieve the effects of DFMO treatment allowing cells to successfully re-enter proliferation [77] contrasting with the irreversible and uniform cellular quiescence in G0/G1 [37] of stumpy cells. The DFMO response therefore does not fully resemble physiological differentiation.
A similar non-physiological response which superficially resembled the natural slender to stumpy differentiation process was observed by Penketh et al. [81] following repeated exposure of cells to low concentrations of the methylating agent 1,2-bis(methylsulfonyl)-1-methylhydrazine. Monomorphic cells treated either in vitro or in vivo morphologically resembled stumpy cells within 48 h, expressed elevated mitochondrial activity and were able to establish stable procyclic cultures when transferred to Cunningham’s Medium. Despite the eventual accumulation of G0/G1 arrested cells upon exposure, analogous to events in natural populations, this was not before progression through a transitional stage that appeared defective in cytokinesis. Specifically, large numbers of multinucleated cells (3–4 nuclei/cell) predominated before semi-synchronous progression to a G0/G1 arrested population [6,81] indicating 1,2-bis(methylsulfonyl)-1-methlhydrazine was not useful as a tool for physiologically-relevant differentiation studies. Cell cycle arrest and morphological transformation to stumpy-like cells has also been observed in vivo following treatment with a small molecule inhibitor of Clan CA cysteine proteinases, carbobenzoxy-phenylalanyl-alanine-diazomethyl ketone (Z-Phe-Ala-CHN2) [82]. Though pleomorphic cells arrested in their growth (without an accumulation of multinucleated cells) and displayed stumpy-like morphology following treatment with the inhibitor, this was accompanied by a significant enlargement of both the lysosome and the parasite, and decreased protein degradation, suggesting that the Z-Phe-Ala-CHN2 response is also non-physiological [82].
An important breakthrough came in 1997 when Vassella et al. described a novel experimental tool which provided insight into the mechanism of stumpy differentiation [6]. Prior to this study, it had been speculated that the slender to stumpy differentiation was triggered by the accumulation of an exogenous growth inhibitor upon reaching a critical cell density, as demonstrated by the ability of plasma from infected animals to inhibit cell proliferation in vitro [83] and the observation that pleomorphic cells would arrest and differentiate to stumpy cells above particular cell densities [6,84]. An elegant series of experiments utilising conditioned medium prepared from bloodstream form cultures at high density demonstrated that parasite density alone induced differentiation, thus excluding cellular programming, depletion of essential nutrients or cell-to-cell contact as possible initiators of the slender to stumpy transition. Pleomorphic cells cultured in the presence of conditioned medium rapidly and uniformly arrested in G0/G1 (without accumulating aberrant cell cycle configurations), morphologically became stumpy, and exhibited NADH diaphorase activity. The effect of the conditioned medium was attributed to a soluble, heat stable, ≤500 Da molecule termed SIF, the identity of which remains elusive [6]. Significantly, SIF-induced differentiated cells were capable of synchronous differentiation to procyclic cells upon a temperature drop and exposure to cis-aconitate, further demonstrating the physiological relevance of the differentiation events induced by exposure to conditioned medium. Interestingly, laboratory-adapted monomorphic cells, incapable of natural differentiation to stumpy cells in vivo, were less responsive to SIF; however conditioned medium prepared from these cell lines was equally as effective as that from pleomorphic cells at inducing cell cycle arrest and differentiation to stumpy cells in differentiation-competent recipients. As such, this suggested that monomorphic cells produce functional SIF, but are in some form defective in either receiving or transducing the SIF signal through a competent pathway [6].
The SIF response pathway represented the next piece in the puzzle. Intracellular levels of cyclic 3′, 5′-adenosine monophosphate (cAMP) had provoked interest following the demonstration of its presence and fluctuation during the course of in vivo slender to stumpy differentiation [85,86]. During infection, the intracellular cAMP content of parasites was shown to increase over three-fold as the parasitaemia reached peak density, then returned to basal levels as stumpy cells accumulated [87]. Hypothesizing a model whereby the density-dependent production of SIF triggers the cAMP signalling pathway to induce differentiation, Vassella et al. [6] exposed pleomorphic cultured cells to the membrane-permeable cAMP analogue 8-(4-chlorophenylthio)-cAMP (8-pCPTcAMP). This generated rapid cell cycle arrest, induction of stumpy morphology, activation of the mitochondrion and accumulation of cells in G0/G1 with near identical kinetics to that of SIF-induced differentiation, with no cytotoxicity. Moreover, intracellular cAMP levels were shown to increase 2–3-fold upon incubation of cells with SIF [6]. Thus, the cAMP signalling pathway was considered a conceivable inducer of slender to stumpy differentiation which responded to the density-mediated accumulation of SIF. The capacity for 8-pCPTcAMP to induce differentiation events in monomorphic cell lines [88] provided further support for the differentiation promoting properties of cAMP, and led to the hypothesis that 8-pCPTcAMP could be a useful tool to identify the genes involved in slender to stumpy differentiation.
Whilst there was little doubt that 8-pCPTcAMP could trigger differentiation events analogous to those initiated by SIF, the perception that cAMP played an important role in the induction and regulation of slender to stumpy differentiation remained in question, as no direct cAMP-dependent effector proteins were annotated in the genome [89]. Ultimately, Laxman et al. [89] were able to demonstrate that the events observed by Vassella et al. [6] were attributable to the potent activity of the hydrolysis product of 8-pCPTcAMP (8-pCPT-2′-O-Me-5′-AMP) rather than 8-pCPTcAMP itself. This was achieved by comparing the effects of membrane-permeable and hydrolysis-resistant 8-pCPTcAMP analogues on cell proliferation [89]. Significantly, this demonstrated that cAMP itself was not involved in the induction of the slender to stumpy differentiation pathway, but rather a hydrolysis product of it, such as AMP.
For differentiation or infection studies to be physiologically relevant, pleomorphic cell culture and genetic manipulation methods required development and optimization. The first report of successful continuous in vitro culture of a pleomorphic cell line came in 1979 when Brun et al. used a mammalian feeder cell layer consisting of a rabbit fibroblast-like cell line grown in modified MEM and supplemented with rabbit serum [90]. Today, the standard method of pleomorphic cell culture involves maintenance of cells densities < 1 × 106 cells/mL [91] for minimal periods of time in HMI9, a medium based on Iscoves modified DMEM medium supplemented with bathocuproinedisulphonic acid, L-cysteine, hypoxanthine, 2-mercaptoethanol, pyruvate, thymidine and foetal bovine serum [92]. For many pleomorphic cell lines the provision of methyl cellulose also assists prolonged culture [84]. Building on the progress in establishing protocols for efficient procyclic [93] and monomorphic [94,95] transfection, MacGregor et al. demonstrated the usefulness of Amaxa Nucleofector technology when applied to pleomorphic cell transfection [91]. Shown to increase the transfection efficiency of monomorphic cells 1000-fold [95], this technology increased the transfection efficiency of pleomorphic AnTat 1.1 90:13 cells [26] to 10−7–10−6 thus permitting routine genetic manipulation of cells capable of a full life cycle progression.
With advances in the repertoire of genetic experimental tools available to study T. brucei, for example the discovery of an active RNAi pathway [96], improved transfection efficiencies [95,97] and the publication of the Tritryp T. brucei genome in 2005 [98], it became possible to perform high throughput gene function analyses. In particular, Alsford et al. described a novel, high resolution experimental approach termed RNA Interference Target Sequencing (RITseq) which would allow for genome-wide analysis of fitness upon induction of RNAi-mediated gene knockdown [36]. An inducible RNAi library, providing around five-fold T. brucei genome coverage [99] was targeted to a unique tagged rRNA locus [97] with an inducible meganuclease cleavage site [36,94]. Cell growth under RNAi-induction led to a loss of representation of target sequences within the remaining proliferating population when RNAi-mediated gene knockdown conferred a fitness deficit. Reads from Illumina sequencing of PCR-amplified RNAi cassette insert DNA could be mapped back to the genome, identifying “cold spots” representative of areas of the genome which were essential for growth in the surveyed assay conditions [36]. Since its description, RITseq has proven to be a phenomenally effective tool in both functional analyses and drug discovery settings. Essential genes for various life cycle stages and events, mechanisms of antitrypanosomal drug action and resistance, and mitochondrial proteins essential for normal cell growth and division have all been identified using RITseq [36,100,101].
4. Molecular Components of the Slender to Stumpy Regulatory Pathway
By combining RITseq technology and selection using differentiation-inducing 8-pCPTcAMP/AMP, Mony et al. were able to identify and validate many intracellular components of the signalling pathway driving stumpy formation [102]. Monomorphic T. brucei populations, transfected with the RNAi library, were exposed to membrane permeable 8-pCPTcAMP/AMP and grown in vitro with or without RNAi induction. Uninduced populations became non-proliferative and underwent cell death in response to 8-pCPTcAMP/AMP. In contrast, the induced population parasites, where components of the signalling pathway had been silenced by RNAi, continued to proliferate in the presence of the differentiation signals. Thereafter, RNAi insert DNA was amplified from the 8-pCPTcAMP/AMP resistant, differentiation defective populations and subjected to Ion Torrent deep sequencing, with reads aligned to the trypanosome genome. The screen identified an extensive cascade of molecules, together assembling a complex signalling pathway driving slender to stumpy differentiation. Groups of identified genes formed distinct tiers of a signalling pathway, from protein kinase and phosphatase signal transducers (e.g., YAK (Tb927.10.15020) and PP1 (Tb927.4.3620/30/40)) to RNA-binding effector molecules such as RBP7 (Tb927.10.12100) [102,103]. Critically, whilst the RNAi library cell line used in the screen to identify pathway components was monomorphic, the physiological relevance of the genes identified by Mony et al. [102] was verified in pleomorphic cell lines. Thus, nine of the genes were tested for sensitivity to SIF upon RNAi induction in vivo. In contrast to the AnTat 1.1 90:13 parental cell line which underwent cell cycle arrest and differentiation to transmission competent stumpy cells, all but two of the induced RNAi lines generated in the study (ADSL (Tb927.9.7550) and ADSS (Tb927.11.3650)) showed increased resistance to SIF, revealed by a delayed or defective differentiation phenotype. Thus, induced cell populations retained slender morphology, showed reduced expression of the stumpy marker PAD1, did not enrich in G0/G1 arrested cells and lacked an elaborated mitochondrion. Mony et al. [102] were therefore able to utilize a potent combination of classic and newly developed technologies and tools to provide information on the genes required to differentiate from proliferative long slender bloodstream forms to transmission competent quiescent stumpy cells [103].
In addition to the aforementioned signal transducers YAK (Tb927.10.15020) and PP1 (Tb927.4.3620/30/40), the α2 subunit of AMPK (Tb927.3.4560) was identified by Mony et al. [102] in the genome-wide RNAi screen. AMPK, a fundamental regulator of energy homeostasis, is activated in response to alterations in the cellular AMP/ATP ratio. In mammalian cells grown in stress conditions, a drop in ATP levels, and a corresponding rise in cellular AMP, triggers AMPK to phosphorylate downstream targets, thus coordinating ATP generating and consuming processes to bring about energy balance [103,104]. Mammalian AMPK is an inhibitor of the evolutionarily conserved protein kinase target of rapamycin (mTOR), a regulator of cell growth, cell cycle, stress response and energy balance [105,106]. Characterisation of the trypanosome TOR like proteins, TbTOR1 and TbTOR2, revealed central functions, analogous to those of their mammalian counterparts, in protein synthesis and cell growth (TbTOR1) and in maintaining cytokinesis (TbTOR2) [107]. Interestingly, however, the structurally and functionally distinct TbTOR4 paralog was described to function as part of a larger complex with TbLST8, MVP and TbArmtor (TbTORC4) to negatively regulate slender to stumpy differentiation [106], thus forming a potential link to the identified AMPK in the Mony et al. screen [102]. Upon induction of TbTOR4 RNAi in monomorphic cultures, cells exhibited hallmarks of slender to stumpy differentiation: morphological change; an irreversible accumulation of cells in G0/G1; mitochondrial elaboration; and the ability to establish procyclic cultures upon treatment with cis-aconitate. Upon TbTOR4 knockdown, mRNA and protein analysis showed a subtle increase in PAD1 expression, though not to the same extent as wild type pleomorphic cells undergoing differentiation. Treatment with 8-pCPTAMP also led to a rapid decrease in TbTOR4 protein levels, supporting the argument against a cAMP dependent pathway and establishing a model where high levels of cellular AMP could activate AMPK which in turn would inhibit the negative regulator TbTOR4, thus allowing the developmental switch from slender to stumpy cells to occur [106]. In addition to TbTOR4, further negative regulators of slender to stumpy form differentiation have been identified, such as MAPK5 [108] and a zinc-finger kinase, ZFK [109]. Though a ZFK null mutant demonstrated reduced growth and increased expression of stumpy markers in vitro the phenotype was not observed in vivo [109]. A MAPK5 null mutant, however, prematurely differentiated to stumpy cells in vivo [108].
More recently, Saldivia et al. [15] have suggested that AMPKα1, and not the AMPKα2 that was identified in the Mony et al. screen [102], is associated with the positive regulation of stumpy form development. In vitro phosphorylation analysis using an α-phospho-Thr172 antibody showed that AMPKα1 phosphorylation was significantly upregulated upon treatment with 8-pCPTAMP, whereas AMPKα2 phosphorylation levels were unaffected. In addition, upon AMPKα1 phosphorylation, levels of stumpy specific transcripts, such as PAD1 and PAD2, were increased and TbTOR4 protein levels were decreased. Moreover, during in vivo analysis with a pleomorphic strain, AMPKα1 was not found be phosphorylated in slender form parasites, whereas its phosphorylation was detected early during differentiation and this was maintained until the population had fully differentiated to stumpy cells. A role for AMPKα1 was further supported by the observation that its phosphorylation was abrogated upon treatment of mice with the AMPK inhibitor compound C, although the specificity of the inhibitor activity was not proven.
5. Future Considerations
With improved molecular descriptions of T. brucei stumpy cells and tools to disrupt or stimulate the differentiation step from slender to stumpy cells, we are now well placed to investigate the signals contributing to T. brucei differentiation in the mammalian host. The availability of cell lines able to report on stumpy formation through PAD1 3′UTR control [28], for example encoding a GFP reporter with a nuclear localisation signal and PAD1 3′UTR [34,58,59], or the use of a PAD1 antibody [27] enables the quantification of stumpy cells in a population. Furthermore, knowledge of the signalling components regulating differentiation [102] has made possible the generation of signal-blind cell lines which do not respond to the quorum-sensing signal. Together such tools could help to identify signals triggering slender to stumpy differentiation through the quorum-sensing signalling pathway, perhaps guiding the identification of the elusive SIF. Regardless of this, the progress made in understanding the molecular control of differentiation in the mammalian bloodstream has revealed new research opportunities and therapeutic implications, as detailed below.
Firstly, understanding of the mechanisms coordinating slender to stumpy differentiation could be employed to force parasites to differentiate prematurely allowing clearance by the host. For example, a differentiation-mediated therapeutic option has already been considered, with compounds capable of inducing slender to stumpy [33] or slender to procyclic form differentiation [110] having been identified in screens using either PAD1 or procyclin 3′UTR regulated reporter cell lines. With identification of the core molecular regulators of differentiation, more targeted therapeutic approaches may now be explored.
Secondly, given the differential sensitivity of slender and stumpy bloodstream parasites to immune attack [55], mild-acid and proteases [14], it is possible that the different biology of stumpy cells makes them more resistant to trypanocidal drugs than slender cells. In the case of Plasmodium falciparum, resistance to the antimalarial drug artemisinin was attributed to persister populations generated by the cell cycle arrest of ring stage parasites, with such parasites able to resume development following removal of drug pressure [111]. The difference in the case of T. brucei is that stumpy cells are irreversibly arrested in their cell cycle and cannot resume development in the bloodstream. Therefore, a drug treatment that killed slender cells but left surviving stumpy cells would not result in recrudescence of the parasites although transmission might still be possible in the short term. Nevertheless, the metabolism or drug sensitivity of monomorphic cells may not fully reflect the sensitivity or resistance of pleomorphic parasites and new therapeutics should be screened on field-relevant strains rather than laboratory adapted lines.
Thirdly, it will be necessary to understand the development from slender to stumpy cells in the context of different mammalian hosts. For example, although stumpy cells were observed to be dominant in T. brucei chronic infection of mice [8], the differentiation status of T. brucei during chronic cattle infection, where parasitaemias are often lower, has not been carefully quantitated. Hence, fly infectivity in acute and chronic stages of infections [112] should be correlated with the proportion and abundance of slender and stumpy cells to determine the relevance of development for the epidemiology of animal African trypanosomiasis (AAT).
Finally, Trypanosoma brucei bloodstream forms can invade beyond the bloodstream and into tissues. In late stages of infection parasites can be found within the brain parenchyma [113]. Recently, other reservoirs of T. brucei have been described including the adipose tissue [34] and the skin [114,115]. Adipose tissue forms of T. brucei were shown to have up-regulated expression of genes involved in fatty acid metabolism, indicating an adaptation to that tissue environment. Additionally, trypanosomes inoculated into skin by tsetse fly bite were found to proliferate within the dermis with close interactions between adipocytes and trypanosomes observed by scanning electron microscopy [115]. In both cases, stumpy cells were identified in addition to those in the circulating bloodstream. Hence, trypanosomes resident in the skin or adipose tissue may contribute to transmission in cases where parasitaemia is very low, as can be the case in chronic infections of sylvatic reservoirs. A further consideration is that the nature of different tissue compartments may affect the diffusion and accumulation of a quorum-sensing signal involved in density-dependent differentiation. Thus, it remains to be determined whether transmissible cells have migrated into the tissue following differentiation in the bloodstream or have undergone differentiation within the tissue. Likewise, the extent of flux between compartments throughout the duration of the infection is currently unknown.
6. Conclusions
The development of T. brucei in the mammalian host is likely coordinated through a complex network of interactions with quorum-sensing mediated density control modulated by different host environments whether this be different host species or different compartments within one host. Moreover, in field settings, the chronic nature of African trypanosome infections often results in co-infections between T. brucei and the other African trypanosome species, Trypanosoma congolense and Trypanosoma vivax [116,117,118]. Thus, the presence of a competitor species of African trypanosome could also contribute to the T. brucei differentiation dynamic in the field. We now have a more detailed picture of the biology and molecular regulation of differentiation in the host bloodstream. Future research will need to build on these discoveries and consider how they apply to the economically important livestock infections and how they might impact on the zoonotic potential of human infective parasites in reservoir hosts. The detailed dissection of the signals mediating trypanosome differentiation within the mammalian host may be challenging, but will provide fascinating insight into the intricate biology tying these parasites to their host, and potentially cohabiting species. This in turn may suggest innovative interventions capable of combatting these persistent pathogens.
Acknowledgments
Work in Keith Matthews’ laboratory is funded by an Investigator Award from the Wellcome Trust (103740/Z/14/Z) and Royal Society Wolfson Research merit award (WM140045) to K.R.M. E.S. is supported by the Wellcome Trust; K.R.McW. is supported by a Medical Research Council UK doctoral studentship. The Centre for Immunity, Infection and Evolution is supported by a Strategic Award from the Wellcome Trust (095831).
Author Contributions
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dyer, N.A.; Rose, C.; Ejeh, N.O.; Acosta-Serrano, A. Flying tryps: Survival and maturation of trypanosomes in tsetse flies. Trends Parasitol. 2013, 29, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Vickerman, K. Antigenic variation in trypanosomes. Nature 1978, 273, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Fenn, K.; Matthews, K.R. The cell biology of Trypanosoma brucei differentiation. Curr. Opin. Microbiol. 2007, 10, 539–546. [Google Scholar] [CrossRef] [PubMed]
- MacGregor, P.; Szoor, B.; Savill, N.J.; Matthews, K.R. Trypanosomal immune evasion, chronicity and transmission: An elegant balancing act. Nat. Rev. Microbiol. 2012, 10, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Matthews, K.R.; McCulloch, R.; Morrison, L.J. The within-host dynamics of African trypanosome infections. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370. [Google Scholar] [CrossRef] [PubMed]
- Vassella, E.; Reuner, B.; Yutzy, B.; Boshart, M. Differentiation of African trypanosomes is controlled by a density sensing mechanism which signals cell cycle arrest via the cAMP pathway. J. Cell Sci. 1997, 110, 2661–2671. [Google Scholar] [PubMed]
- Reuner, B.; Vassella, E.; Yutzy, B.; Boshart, M. Cell density triggers slender to stumpy differentiation of Trypanosoma brucei bloodstream forms in culture. Mol. Biochem. Parasitol. 1997, 90, 269–280. [Google Scholar] [CrossRef]
- MacGregor, P.; Savill, N.J.; Hall, D.; Matthews, K.R. Transmission stages dominate trypanosome within-host dynamics during chronic infections. Cell Host Microbe 2011, 9, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, M.T. A comparison between a syringe-passaged and a tsetse-fly-transmitted line of a strain of Trypanosoma rhodesiense. Ann. Trop. Med. Parasitol. 1960, 54, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Bruce, D.; Harvey, D.; Hamerton, A.E.; Davey, J.B.; Bruce, L. The morphology of the trypanosome causing disease in man in Nyasaland. Proc. R. Soc. Lond. Ser. B Contain. Pap. Biol. Character 1912, 85, 423. [Google Scholar] [CrossRef]
- Robertson, M. Notes on the polymorphism of Trypanosoma gambiense in the blood and its relation to the exogenous cycle in Glossina palpalis. Proc. R. Soc. Lond. Ser. B Contain. Pap. Biol. Character 1912, 85, 527. [Google Scholar] [CrossRef]
- Wijers, D.J.; Willett, K.C. Factors that may influence the infection rate of Glossina palpalis with Trypanosoma gambiense. Ii. The number and morphology of the trypanosomes present in the blood of the host at the time of the infected feed. Ann. Trop. Med. Parasitol. 1960, 54, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Turner, C.; Barry, J.; Vickerman, K. Loss of variable antigen during transformation of Trypanosoma brucei rhodesiense from bloodstream to procyclic forms in the tsetse fly. Parasitol. Res. 1988, 74, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Nolan, D.P.; Rolin, S.; Rodriguez, J.R.; van Den Abbeele, J.; Pays, E. Slender and stumpy bloodstream forms of Trypanosoma brucei display a differential response to extracellular acidic and proteolytic stress. Eur. J. Biochem. 2000, 267, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Saldivia, M.; Ceballos-Pérez, G.; Bart, J.-M.; Navarro, M. The AMPKα1 pathway positively regulates the developmental transition from proliferation to quiescence in Trypanosoma brucei. Cell Rep. 2016, 17, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Sbicego, S.; Vassella, E.; Kurath, U.; Blum, B.; Roditi, I. The use of transgenic Trypanosoma brucei to identify compounds inducing the differentiation of bloodstream forms to procyclic forms. Mol. Biochem. Parasitol. 1999, 104, 311–322. [Google Scholar] [CrossRef]
- Yabu, Y.; Takayanagi, T. Trypsin-stimulated transformation of Trypanosoma brucei gambiense bloodstream forms to procyclic forms in vitro. Parasitol. Res. 1988, 74, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Szöőr, B.; Dyer, N.A.; Ruberto, I.; Acosta-Serrano, A.; Matthews, K.R. Independent pathways can transduce the life-cycle differentiation signal in Trypanosoma brucei. PLoS Pathog. 2013, 9, e1003689. [Google Scholar] [CrossRef] [PubMed]
- Rolin, S.; Hancocq-Quertier, J.; Paturiaux-Hanocq, F.; Nolan, D.P.; Pays, E. Mild acid stress as a differentiation trigger in Trypanosoma brucei. Mol. Biochem. Parasitol. 1998, 93, 251–262. [Google Scholar] [CrossRef]
- Bursell, E. Aspects of the metabolism of amino acids in the tsetse fly, Glossina (diptera). J. Insect Physiol. 1963, 9, 439–452. [Google Scholar] [CrossRef]
- Mantilla, B.S.; Marchese, L.; Casas–Sánchez, A.; Dyer, N.A.; Ejeh, N.; Biran, M.; Bringaud, F.; Lehane, M.J.; Acosta-Serrano, A.; Silber, A.M. Proline Metabolism is Essential for Trypanosoma brucei brucei Survival in the Tsetse Vector. PLoS Pathog. 2017, 13, e1006158. [Google Scholar] [CrossRef] [PubMed]
- Vickerman, K. Developmental cycles and biology of pathogenic trypanosomes. Br. Med. Bull. 1985, 41, 105. [Google Scholar] [CrossRef] [PubMed]
- Vickerman, K. Polymorphism and mitochondrial activity in sleeping sickness trypanosomes. Nature 1965, 208, 762. [Google Scholar] [CrossRef] [PubMed]
- Vassella, E.; Straesser, K.; Boshart, M. A mitochondrion-specific dye for multicolour fluorescent imaging of Trypanosoma brucei. Mol. Biochem. Parasitol. 1997, 90, 381–385. [Google Scholar] [CrossRef]
- Vanhollebeke, B.; Uzureau, P.; Monteyne, D.; Perez-Morga, D.; Pays, E. Cellular and molecular remodeling of the endocytic pathway during differentiation of Trypanosoma brucei bloodstream forms. Eukaryot. Cell 2010, 9, 1272–1282. [Google Scholar] [CrossRef] [PubMed]
- Engstler, M.; Boshart, M. Cold shock and regulation of surface protein trafficking convey sensitization to inducers of stage differentiation in Trypanosoma brucei. Genes Dev. 2004, 18, 2798. [Google Scholar] [CrossRef] [PubMed]
- Dean, S.; Marchetti, R.; Kirk, K.; Matthews, K.R. A surface transporter family conveys the trypanosome differentiation signal. Nature 2009, 459, 213–217. [Google Scholar] [CrossRef] [PubMed]
- MacGregor, P.; Matthews, K.R. Identification of the regulatory elements controlling the transmission stage-specific gene expression of PAD1 in Trypanosoma brucei. Nucleic Acids Res. 2012, 40, 7705–7717. [Google Scholar] [CrossRef] [PubMed]
- Barnwell, E.M.; van Deursen, F.J.; Jeacock, L.; Smith, K.A.; Maizels, R.M.; Acosta–Serrano, A.; Matthews, K. Developmental regulation and extracellular release of a VSG expression–site–associated gene product from Trypanosoma brucei bloodstream forms. J. Cell Sci. 2010, 123, 3401–3411. [Google Scholar] [CrossRef] [PubMed]
- Florent, I.C.; Raibaud, A.; Eisen, H. A family of genes related to a new expression site-associated gene in Trypanosoma equiperdum. Mol. Cell. Biol. 1991, 11, 2180–2188. [Google Scholar] [CrossRef] [PubMed]
- Hertz-Fowler, C.; Figueiredo, L.M.; Quail, M.A.; Becker, M.; Jackson, A.; Bason, N.; Brooks, K.; Churcher, C.; Fahkro, S.; Goodhead, I.; et al. Telomeric expression sites are highly conserved in Trypanosoma brucei. PLoS ONE 2008, 3, e3527. [Google Scholar] [CrossRef] [PubMed]
- Monk, S.L.; Simmonds, P.; Matthews, K.R. A short bifunctional element operates to positively or negatively regulate ESAG9 expression in different developmental forms of Trypanosoma brucei. J. Cell Sci. 2013, 126, 2294. [Google Scholar] [CrossRef] [PubMed]
- MacGregor, P.; Ivens, A.; Shave, S.; Collie, I.; Gray, D.; Auer, M.; Matthews, K.R. High-throughput chemical screening for antivirulence developmental phenotypes in Trypanosoma brucei. Eukaryot. Cell 2014, 13, 412–426. [Google Scholar] [CrossRef] [PubMed]
- Trindade, S.; Rijo-Ferreira, F.; Carvalho, T.; Pinto-Neves, D.; Guegan, F.; Aresta-Branco, F.; Bento, F.; Young, S.A.; Pinto, A.; van Den Abbeele, J.; et al. Trypanosoma brucei parasites occupy and functionally adapt to the adipose tissue in mice. Cell Host Microbe 2016, 19, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Rico, E.; Ivens, A.; Glover, L.; Horn, D.; Matthews, K.R. Genome-wide RNAi selection identifies a regulator of transmission stage-enriched gene families and cell-type differentiation in Trypanosoma brucei. PLoS Pathog. 2017, 13, e1006279. [Google Scholar] [CrossRef] [PubMed]
- Alsford, S.; Turner, D.J.; Obado, S.O.; Sanchez-Flores, A.; Glover, L.; Berriman, M.; Hertz-Fowler, C.; Horn, D. High-throughput phenotyping using parallel sequencing of RNA interference targets in the African trypanosome. Genome Res. 2011, 21, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, S.Z.; Naessens, J.; Liesegang, B.; Moloo, S.K.; Magondu, J. Analysis by flow cytometry of DNA synthesis during the life cycle of African trypanosomes. Acta Trop. 1984, 41, 313–323. [Google Scholar] [PubMed]
- Matthews, K.R.; Gull, K. Evidence for an interplay between cell cycle progression and the initiation of differentiation between life cycle forms of African trypanosomes. J. Cell Biol. 1994, 125, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Sherwin, T.; Ploubidou, A.; Byard, E.H.; Gull, K. Microtubule polarity and dynamics in the control of organelle positioning, segregation, and cytokinesis in the trypanosome cell cycle. J. Cell Biol. 1995, 128, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Matthews, K.R.; Sherwin, T.; Gull, K. Mitochondrial genome repositioning during the differentiation of the African trypanosome between life cycle forms is microtubule mediated. J. Cell Sci. 1995, 108, 2231. [Google Scholar] [PubMed]
- Ziegelbauer, K.; Quinten, M.; Schwarz, H.; Pearson, T.W.; Overath, P. Synchronous differentiation of Trypanosoma brucei from bloodstream to procyclic forms in vitro. Eur. J. Biochem. 1990, 192, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Overath, P.; Czichos, J.; Haas, C. The effect of citrate/cis-aconitate on oxidative metabolism during transformation of Trypanosoma brucei. Eur. J. Biochem. 1986, 160, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Brun, R.; Schonenberger, M. Stimulating effect of citrate and cis-aconitate on the transformation of Trypanosoma brucei bloodstream forms to procyclic forms in vitro. Z. fur Parasitenkund. 1981, 66, 17–24. [Google Scholar] [CrossRef]
- Roditi, I.; Schwarz, H.; Pearson, T.W.; Beecroft, R.P.; Liu, M.K.; Richardson, J.P.; Bühring, H.J.; Pleiss, J.; Bülow, R.; Williams, R.O. Procyclin gene expression and loss of the variant surface glycoprotein during differentiation of Trypanosoma brucei. J. Cell Biol. 1989, 108, 737. [Google Scholar] [CrossRef] [PubMed]
- Overath, P.; Czichos, J.; Stock, U.; Nonnengaesser, C. Repression of glycoprotein synthesis and release of surface coat during transformation of Trypanosoma brucei. EMBO J. 1983, 2, 1721–1728. [Google Scholar] [PubMed]
- Gruszynski, A.E.; van Deursen, F.J.; Albareda, M.C.; Best, A.; Chaudhary, K.; Cliffe, L.J.; del Rio, L.; Dunn, J.D.; Ellis, L.; Evans, K.J.; et al. Regulation of surface coat exchange by differentiating African trypanosomes. Mol. Biochem. Parasitol. 2006, 147, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Matthews, K.R.; Gull, K. Commitment to differentiation and cell cycle re-entry are coincident but separable events in the transformation of African trypanosomes from their bloodstream to their insect form. J. Cell Sci. 1997, 110, 2609. [Google Scholar] [PubMed]
- Szöőr, B.; Wilson, J.; McElhinney, H.; Tabernero, L.; Matthews, K.R. Protein tyrosine phosphatase TbPTP1: A molecular switch controlling life cycle differentiation in trypanosomes. J. Cell Sci. 2006, 175, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Szöőr, B.; Ruberto, I.; Burchmore, R.; Matthews, K.R. A novel phosphatase cascade regulates differentiation in Trypanosoma brucei via a glycosomal signaling pathway. Genes Dev. 2010, 24, 1306. [Google Scholar] [CrossRef] [PubMed]
- Mulindwa, J.; Mercé, C.; Matovu, E.; Enyaru, J.; Clayton, C. Transcriptomes of newly-isolated Trypanosoma brucei rhodesiense reveal hundreds of mRNAs that are co-regulated with stumpy-form markers. BMC Genom. 2015, 16, 1118. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Sananes, M.R.; Szoor, B.; Ferguson, M.A.; Urbaniak, M.D.; Matthews, K.R. Molecular control of irreversible bistability during trypanosome developmental commitment. J. Cell Biol. 2015, 211, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Gale, M.; Carter, V.; Parsons, M. Translational control mediates the developmental regulation of the Trypanosoma brucei NRK protein kinase. J. Biol. Chem. 1994, 269, 31659–31665. [Google Scholar] [PubMed]
- Horn, D. Antigenic variation in African trypanosomes. Mol. Biochem. Parasitol. 2014, 195, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Amiguet-Vercher, A.; Pérez-Morga, D.; Pays, A.; Poelvoorde, P.; van Xong, H.; Tebabi, P.; Vanhamme, L.; Pays, E. Loss of the mono-allelic control of the VSG expression sites during the development of Trypanosoma brucei in the bloodstream. Mol. Microbiol. 2004, 51, 1577–1588. [Google Scholar] [CrossRef] [PubMed]
- Engstler, M.; Pfohl, T.; Herminghaus, S.; Boshart, M.; Wiegertjes, G.; Heddergott, N.; Overath, P. Hydrodynamic flow- mediated protein sorting on the cell surface of trypanosomes. Cell 2007, 131, 505–515. [Google Scholar] [CrossRef] [PubMed]
- McLintock, L.M.; Turner, C.M.; Vickerman, K. Comparison of the effects of immune killing mechanisms on Trypanosoma brucei parasites of slender and stumpy morphology. Parasite Immunol. 1993, 15, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Navarro, M.; Gull, K. A pol I transcriptional body associated with VSG mono-allelic expression in Trypanosoma brucei. Nature 2001, 414, 759. [Google Scholar] [CrossRef] [PubMed]
- Batram, C.; Jones, N.G.; Janzen, C.J.; Markert, S.M.; Engstler, M. Expression site attenuation mechanistically links antigenic variation and development in Trypanosoma brucei. eLife 2014, 3, e02324. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H.; Subota, I.; Batram, C.; Kramer, S.; Janzen, C.J.; Jones, N.G.; Engstler, M. A quorum sensing-independent path to stumpy development in Trypanosoma brucei. PLoS Pathog. 2017, 13, e1006324. [Google Scholar] [CrossRef] [PubMed]
- Brecht, M.; Parsons, M. Changes in polysome profiles accompany trypanosome development. Mol. Biochem. Parasitol. 1998, 97, 189–198. [Google Scholar] [CrossRef]
- Capewell, P.; Monk, S.; Ivens, A.; Macgregor, P.; Fenn, K.; Walrad, P.; Bringaud, F.; Smith, T.K.; Matthews, K.R. Regulation of Trypanosoma brucei total and polysomal mRNA during development within its mammalian host. PLoS ONE 2013, 8, e67069. [Google Scholar] [CrossRef] [PubMed]
- Kabani, S.; Fenn, K.; Ross, A.; Ivens, A.; Smith, T.K.; Ghazal, P.; Matthews, K. Genome-wide expression profiling of in vivo-derived bloodstream parasite stages and dynamic analysis of mRNA alterations during synchronous differentiation in Trypanosoma brucei. BMC Genom. 2009, 10, 427. [Google Scholar] [CrossRef] [PubMed]
- Jensen, B.C.; Sivam, D.; Kifer, C.T.; Myler, P.J.; Parsons, M. Widespread variation in transcript abundance within and across developmental stages of Trypanosoma brucei. BMC Genom. 2009, 10, 482. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, D.; Gunasekera, K.; Mani, J.; Osteras, M.; Farinelli, L.; Baerlocher, L.; Roditi, I.; Ochsenreiter, T. Spliced leader trapping reveals widespread alternative splicing patterns in the highly dynamic transcriptome of Trypanosoma brucei. PLoS Pathog. 2010, 6, e1001037. [Google Scholar] [CrossRef] [PubMed]
- Clayton, C.E. Life without transcriptional control? From fly to man and back again. EMBO J. 2002, 21, 1881–1888. [Google Scholar] [CrossRef] [PubMed]
- Butter, F.; Bucerius, F.; Michel, M.; Cicova, Z.; Mann, M.; Janzen, C.J. Comparative proteomics of two life cycle stages of stable isotope-labeled Trypanosoma brucei reveals novel components of the parasite’s host adaptation machinery. Mol. Cell. Proteom. 2013, 12, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Urbaniak, M.D.; Guther, M.L.S.; Ferguson, M.A.J. Comparative SILAC proteomic analysis of Trypanosoma brucei bloodstream and procyclic lifecycle stages. PLoS ONE 2012, 7, e36619. [Google Scholar] [CrossRef] [PubMed]
- Gunasekera, K.; Wüthrich, D.; Braga-Lagache, S.; Heller, M.; Ochsenreiter, T. Proteome remodelling during development from blood to insect-form Trypanosoma brucei quantified by SILAC and mass spectrometry. BMC Genom. 2012, 13, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Dejung, M.; Subota, I.; Bucerius, F.; Dindar, G.; Freiwald, A.; Engstler, M.; Boshart, M.; Butter, F.; Janzen, C.J. Quantitative proteomics uncovers novel factors involved in developmental differentiation of Trypanosoma brucei. PLoS Pathog. 2016, 12, e1005439. [Google Scholar] [CrossRef] [PubMed]
- Rijo-Ferreira, F.; Pinto-Neves, D.; Barbosa-Morais, N.L.; Takahashi, J.S.; Figueiredo, L.M. Trypanosoma brucei metabolism is under circadian control. Nat. Microbiol. 2017, 2, 17032. [Google Scholar] [CrossRef] [PubMed]
- Sherwin, T.; Gull, K. The cell division cycle of Trypanosoma brucei brucei: Timing of event markers and cytoskeletal modulations. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1989, 323, 573–588. [Google Scholar] [CrossRef] [PubMed]
- Roditi, I.; Carrington, M.; Turner, M. Expression of a polypeptide containing a dipeptide repeat is confined to the insect stage of Trypanosoma brucei. Nature 1987, 325, 272–274. [Google Scholar] [CrossRef] [PubMed]
- Steverding, D. The development of drugs for treatment of sleeping sickness: A historical review. Parasit Vectors 2010, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Willert, E.; Phillips, M.A. Regulation and function of polyamines in African trypanosomes. Trends Parasitol. 2012, 28, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Meyskens, F.L., Jr.; Gerner, E.W. Development of difluoromethylornithine (DFMO) as a chemoprevention agent. Clin. Cancer Res. 1999, 5, 945–951. [Google Scholar] [PubMed]
- Pegg, A.E.; McCann, P.P. Polyamine metabolism and function. Am. J. Physiol. 1982, 243, C212–C221. [Google Scholar] [PubMed]
- Giffin, B.F.; McCann, P.P.; Bitonti, A.J.; Bacchi, C.J. Polyamine depletion following exposure to DL-alpha-difluoromethylornithine both in vivo and in vitro initiates morphological alterations and mitochondrial activation in a monomorphic strain of Trypanosoma brucei brucei. J. Protozool. 1986, 33, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Bacchi, C.J.; Garofalo, J.; Mockenhaupt, D.; McCann, P.P.; Diekema, K.A.; Pegg, A.E.; Nathan, H.C.; Mullaney, E.A.; Chunosoff, L.; Sjoerdsma, A.; et al. In vivo effects of alpha-DL-difluoromethylornithine on the metabolism and morphology of Trypanosoma brucei brucei. Mol. Biochem. Parasitol. 1983, 7, 209–225. [Google Scholar] [CrossRef]
- De Gee, A.L.; Carstens, P.H.; McCann, P.P.; Mansfield, J.M. Morphological changes in Trypanosoma brucei rhodesiense following inhibition of polyamine biosynthesis in vivo. Tissue Cell 1984, 16, 731–738. [Google Scholar] [CrossRef]
- Giffin, B.F.; McCann, P.P. Physiological activation of the mitochondrion and the transformation capacity of DFMO induced intermediate and short stumpy bloodstream form trypanosomes. Am. J. Trop. Med. Hyg. 1989, 40, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Penketh, P.G.; Divo, A.A.; Shyam, K.; Patton, C.L.; Sartorelli, A.C. The effects of the methylating agent 1,2-bis(methylsulfonyl)-1-methylhydrazine on morphology, DNA content and mitochondrial function of Trypanosoma brucei subspecies. J. Protozool. 1991, 38, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Scory, S.; Stierhof, Y.D.; Caffrey, C.R.; Steverding, D. The cysteine proteinase inhibitor Z-PHE-ALA-CHN2 alters cell morphology and cell division activity of Trypanosoma brucei bloodstream forms in vivo. Kinetoplastid Biol. Dis. 2007, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Seed, J.R.; Sechelski, J.B. Mechanism of long slender (LS) to short stumpy (SS) transformation in the African trypanosomes. J. Protozool. 1989, 36, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Vassella, E.; Boshart, M. High molecular mass agarose matrix supports growth of bloodstream forms of pleomorphic Trypanosoma brucei strains in axenic culture. Mol. Biochem. Parasitol. 1996, 82, 91–105. [Google Scholar] [CrossRef]
- Strickler, J.E.; Patton, C.L. Adenosine 3′,5′-monophosphate in reproducing and differentiated trypanosomes. Science 1975, 190, 1110–1112. [Google Scholar] [CrossRef] [PubMed]
- Tagoe, D.N.; Kalejaiye, T.D.; de Koning, H.P. The ever unfolding story of cAMP signaling in trypanosomatids: Vive la difference! Front. Pharmacol. 2015, 6, 185. [Google Scholar] [CrossRef] [PubMed]
- Mancini, P.E.; Patton, C.L. Cyclic 3′,5′-adenosine monophosphate levels during the developmental cycle of Trypanosoma brucei brucei in the rat. Mol. Biochem. Parasitol. 1981, 3, 19–31. [Google Scholar] [CrossRef]
- Breidbach, T.; Ngazoa, E.; Steverding, D. Trypanosoma brucei: In vitro slender-to-stumpy differentiation of culture-adapted, monomorphic bloodstream forms. Exp. Parasitol. 2002, 101, 223–230. [Google Scholar] [CrossRef]
- Laxman, S.; Riechers, A.; Sadilek, M.; Schwede, F.; Beavo, J.A. Hydrolysis products of cAMP analogs cause transformation of Trypanosoma brucei from slender to stumpy-like forms. Proc. Natl. Acad. Sci. USA 2006, 103, 19194–19199. [Google Scholar] [CrossRef] [PubMed]
- Brun, R.; Jenni, L.; Tanner, M.; Schonenberger, M.; Schell, K.F. Cultivation of vertebrate infective forms derived from metacyclic forms of pleomorphic Trypanosoma brucei stocks. Short communication. Acta Trop. 1979, 36, 387–390. [Google Scholar] [PubMed]
- MacGregor, P.; Rojas, F.; Dean, S.; Matthews, K.R. Stable transformation of pleomorphic bloodstream form Trypanosoma brucei. Mol. Biochem. Parasitol. 2013, 190, 60–62. [Google Scholar] [CrossRef] [PubMed]
- Hirumi, H.; Hirumi, K. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 1989, 75, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Van der Ploeg, L.H. Homologous recombination and stable transfection in the parasitic protozoan Trypanosoma brucei. Science 1990, 250, 1583–1587. [Google Scholar] [CrossRef] [PubMed]
- Glover, L.; Horn, D. Site-specific DNA double-strand breaks greatly increase stable transformation efficiency in Trypanosoma brucei. Mol. Biochem. Parasitol. 2009, 166, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Burkard, G.; Fragoso, C.M.; Roditi, I. Highly efficient stable transformation of bloodstream forms of Trypanosoma brucei. Mol. Biochem. Parasitol. 2007, 153, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Ngo, H.; Tschudi, C.; Gull, K.; Ullu, E. Double-stranded RNA induces mRNA degradation in Trypanosoma brucei. Proc. Natl. Acad. Sci. USA 1998, 95, 14687–14692. [Google Scholar] [CrossRef] [PubMed]
- Alsford, S.; Kawahara, T.; Glover, L.; Horn, D. Tagging a T. brucei rRNA locus improves stable transfection efficiency and circumvents inducible expression position effects. Mol. Biochem. Parasitol. 2005, 144, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Berriman, M.; Ghedin, E.; Hertz-Fowler, C.; Blandin, G.; Renauld, H.; Bartholomeu, D.C.; Lennard, N.J.; Caler, E.; Hamlin, N.E.; Haas, B.; et al. The genome of the African trypanosome Trypanosoma brucei. Science 2005, 309, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.C.; Wang, Z.; Drew, M.E.; Englund, P.T. Glycolysis modulates trypanosome glycoprotein expression as revealed by an RNAi library. EMBO J. 2002, 21, 4429–4438. [Google Scholar] [CrossRef] [PubMed]
- Alsford, S.; Eckert, S.; Baker, N.; Glover, L.; Sanchez-Flores, A.; Leung, K.F.; Turner, D.J.; Field, M.C.; Berriman, M.; Horn, D. High-throughput decoding of antitrypanosomal drug efficacy and resistance. Nature 2012, 482, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Mbang-Benet, D.E.; Sterkers, Y.; Crobu, L.; Sarrazin, A.; Bastien, P.; Pages, M. RNA interference screen reveals a high proportion of mitochondrial proteins essential for correct cell cycle progress in Trypanosoma brucei. BMC Genom. 2015, 16, 297. [Google Scholar] [CrossRef] [PubMed]
- Mony, B.M.; MacGregor, P.; Ivens, A.; Rojas, F.; Cowton, A.; Young, J.; Horn, D.; Matthews, K. Genome wide dissection of the quorum sensing signaling pathway in Trypanosoma brucei. Nature 2014, 505, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Mony, B.M.; Matthews, K.R. Assembling the components of the quorum sensing pathway in African trypanosomes. Mol. Microbiol. 2015, 96, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Novikova, D.S.; Garabadzhiu, A.V.; Melino, G.; Barlev, N.A.; Tribulovich, V.G. AMP-activated protein kinase: Structure, function, and role in pathological processes. Biochemistry 2015, 80, 127–144. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Kim, J.; Guan, K.L. AMPK and mTOR in cellular energy homeostasis and drug targets. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 381–400. [Google Scholar] [CrossRef] [PubMed]
- Barquilla, A.; Saldivia, M.; Diaz, R.; Bart, J.M.; Vidal, I.; Calvo, E.; Hall, M.N.; Navarro, M. Third target of rapamycin complex negatively regulates development of quiescence in Trypanosoma brucei. Proc. Natl. Acad. Sci. USA 2012, 109, 14399–14404. [Google Scholar] [CrossRef] [PubMed]
- Barquilla, A.; Crespo, J.L.; Navarro, M. Rapamycin inhibits trypanosome cell growth by preventing TOR complex 2 formation. Proc. Natl. Acad. Sci. USA 2008, 105, 14579–14584. [Google Scholar] [CrossRef] [PubMed]
- Domenicali Pfister, D.; Burkard, G.; Morand, S.; Renggli, C.K.; Roditi, I.; Vassella, E. A mitogen-activated protein kinase controls differentiation of bloodstream forms of Trypanosoma brucei. Eukaryot. Cell 2006, 5, 1126. [Google Scholar] [CrossRef] [PubMed]
- Vassella, E.; Krämer, R.; Turner, C.M.; Wankell, M.; Modes, C.; van den Bogaard, M.; Boshart, M. Deletion of a novel protein kinase with PX and FYVE-related domains increases the rate of differentiation of Trypanosoma brucei. Mol. Microbiol. 2001, 41, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Wenzler, T.; Schumann Burkard, G.; Schmidt, R.S.; Mäser, P.; Bergner, A.; Roditi, I.; Brun, R. A new approach to chemotherapy: Drug–induced differentiation kills African trypanosomes. Sci. Rep. 2016, 6, 22451. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, B.; Lelievre, J.; Lopez Barragan, M.J.; Laurent, V.; Su, X.-Z.; Berry, A.; Benoit-Vical, F. Increased tolerance to artemisinin in Plasmodium falciparum is mediated by a quiescence mechanism. Antimicrob. Agents Chemother. 2010, 54, 1872. [Google Scholar] [CrossRef] [PubMed]
- Van Den Bossche, P.; Ky-zerbo, A.; Brandt, J.; Marcotty, T.; Geerts, S.; de Deken, R. Transmissibility of Trypanosoma brucei during its development in cattle. Trop. Med. Int. Health 2005, 10, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Masocha, W.; Rottenberg, M.E.; Kristensson, K. Migration of African trypanosomes across the blood-brain barrier. Physiol. Behav. 2007, 92, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Capewell, P.; Cren-Travaillé, C.; Marchesi, F.; Johnston, P.; Clucas, C.; Benson, R.A.; Gorman, T.-A.; Calvo-Alvarez, E.; Crouzols, A.; Jouvion, G.; et al. The skin is a significant but overlooked anatomical reservoir for vector-borne African trypanosomes. eLife 2016, 5, e17716. [Google Scholar] [CrossRef] [PubMed]
- Caljon, G.; van Reet, N.; De Trez, C.; Vermeersch, M.; Pérez-Morga, D.; van Den Abbeele, J. The dermis as a delivery site of Trypanosoma brucei for tsetse flies. PLoS Pathog. 2016, 12, e1005744. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.P.; Tosas, O.; Tilley, A.; Picozzi, K.; Coleman, P.; Hide, G.; Welburn, S.C. Constraints to estimating the prevalence of trypanosome infections in East African zebu cattle. Parasites Vectors 2010, 3, 210. [Google Scholar] [CrossRef] [PubMed]
- Thumbi, S.M.; Jung’a, J.O.; Mosi, R.O.; McOdimba, F.A. Spatial distribution of African animal trypanosomiasis in Suba and Teso districts in Western Kenya. BMC Res. Notes 2010, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.E.; Mubanga, J.; Fevre, E.M.; Picozzi, K.; Eisler, M.C.; Thomas, R.; Welburn, S.C. Characterisation of the wildlife reservoir community for human and animal trypanosomiasis in the Luangwa Valley, Zambia (HAT and nagana in wildlife in Zambia). PLoS Negl. Trop. Dis. 2011, 5, e1211. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).