
Modeling HSV-1 Latency in Human Embryonic Stem Cell-Derived Neurons



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
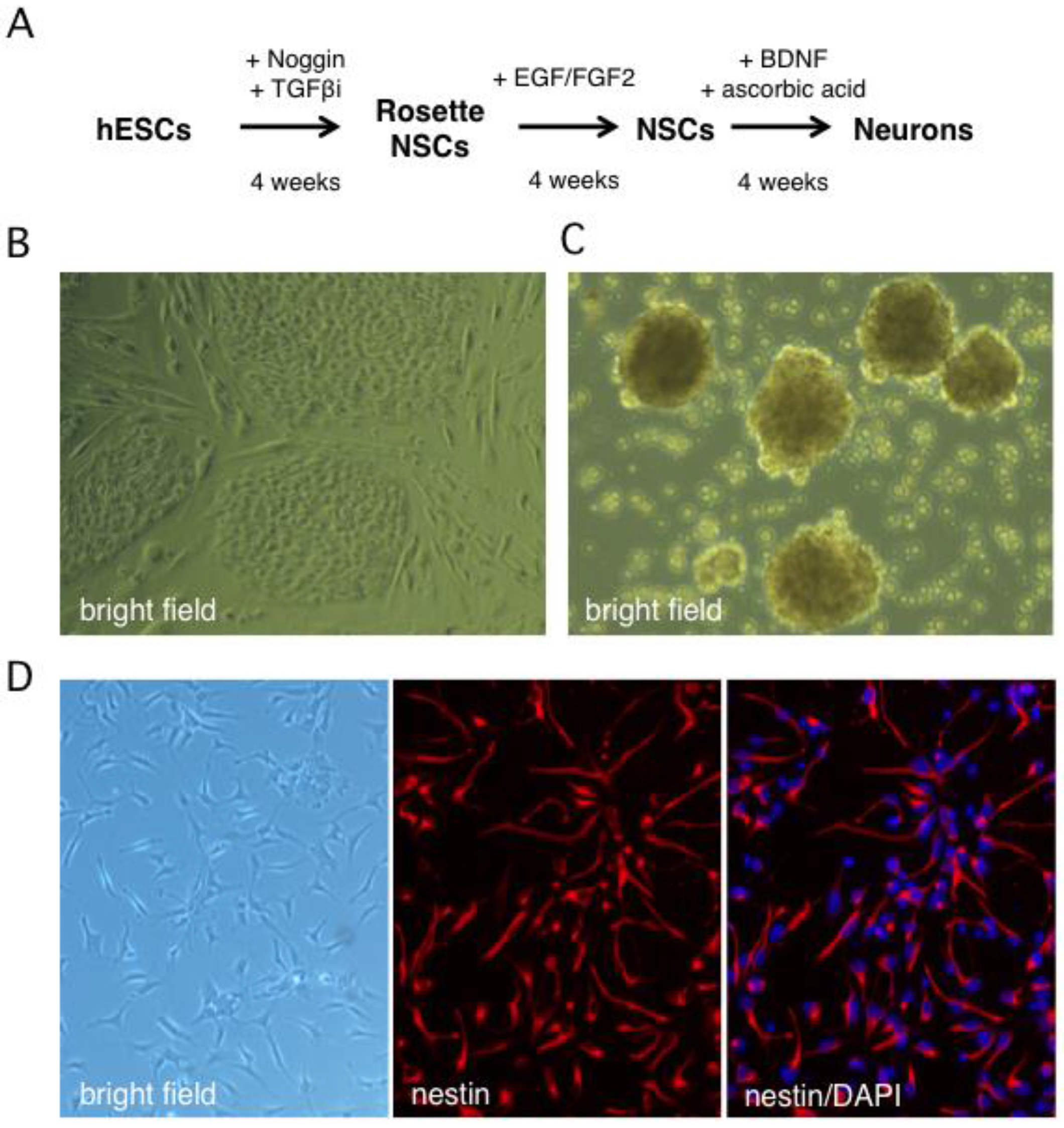
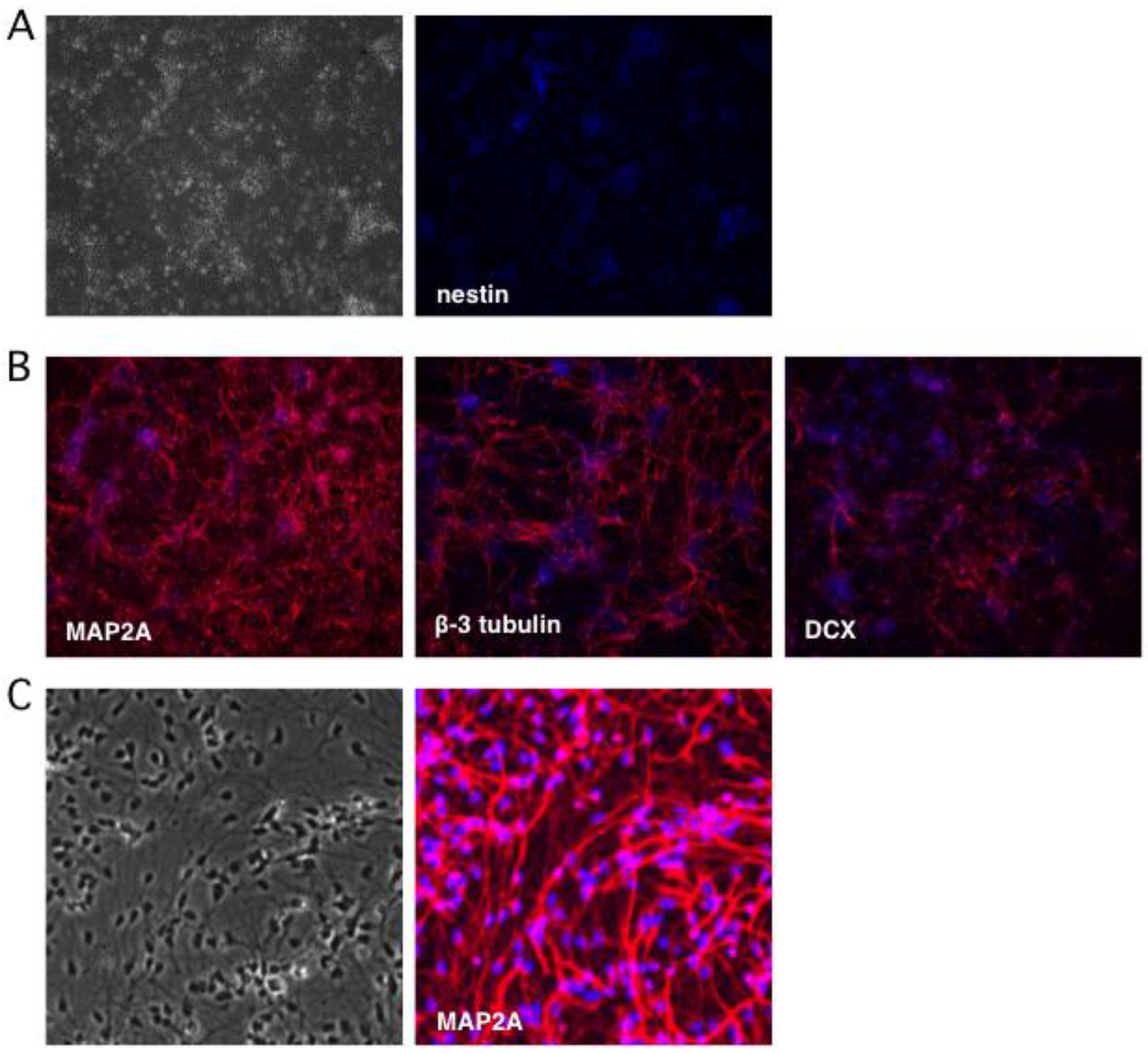
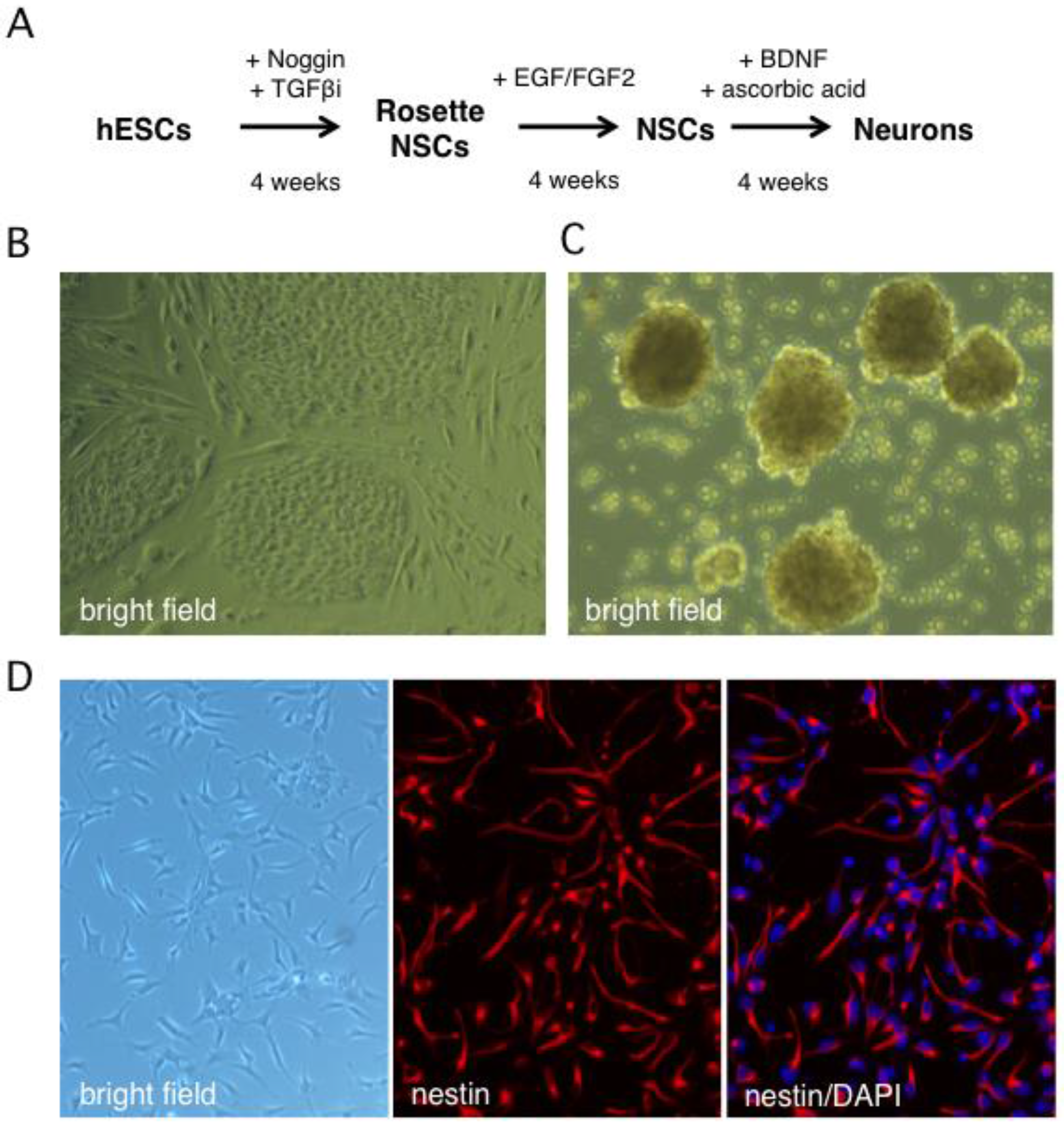
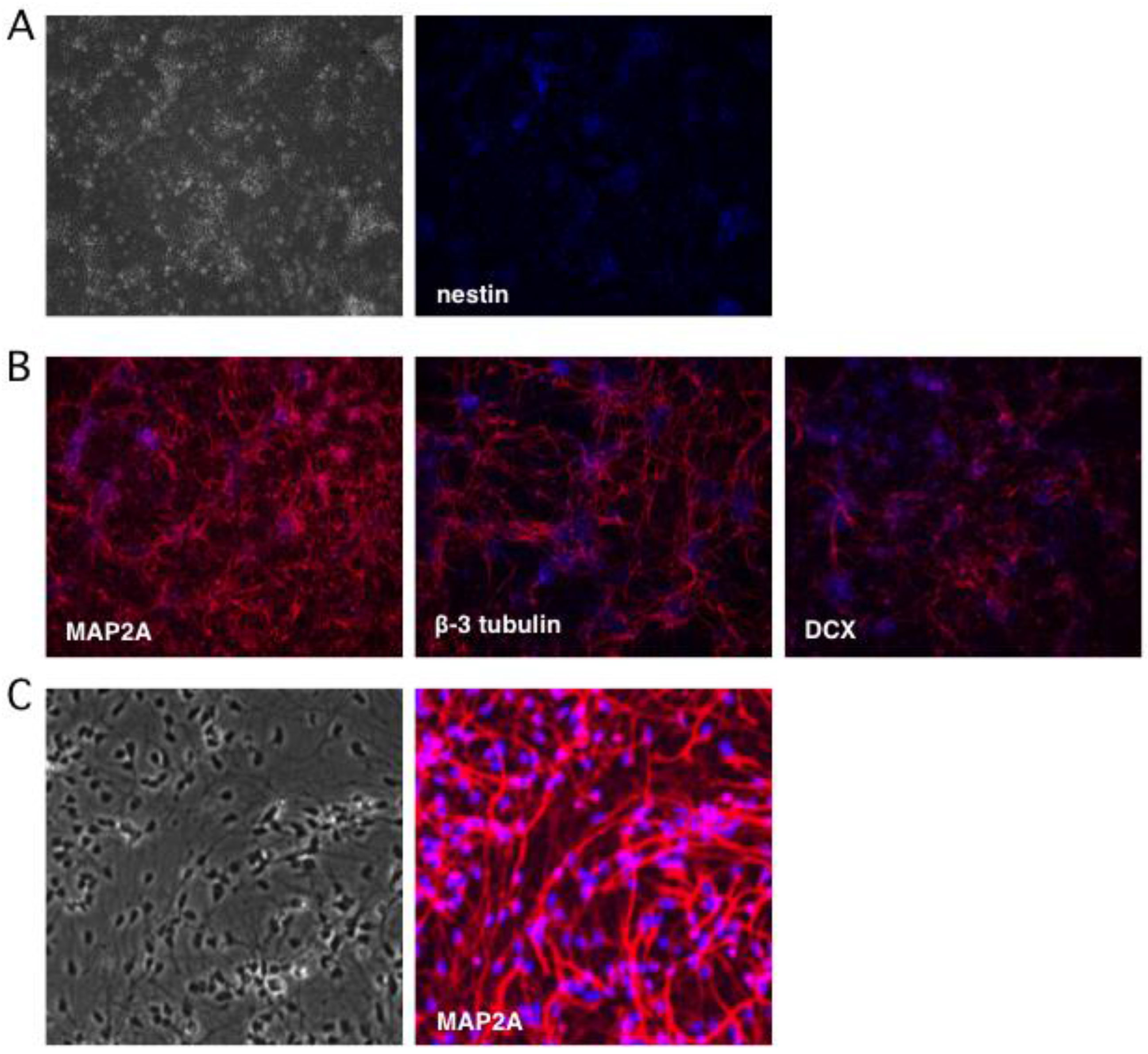
2.1. Stepwise Differentiation of the Hes5::GFP Human Embryonic Stem Cell Line into Neurons
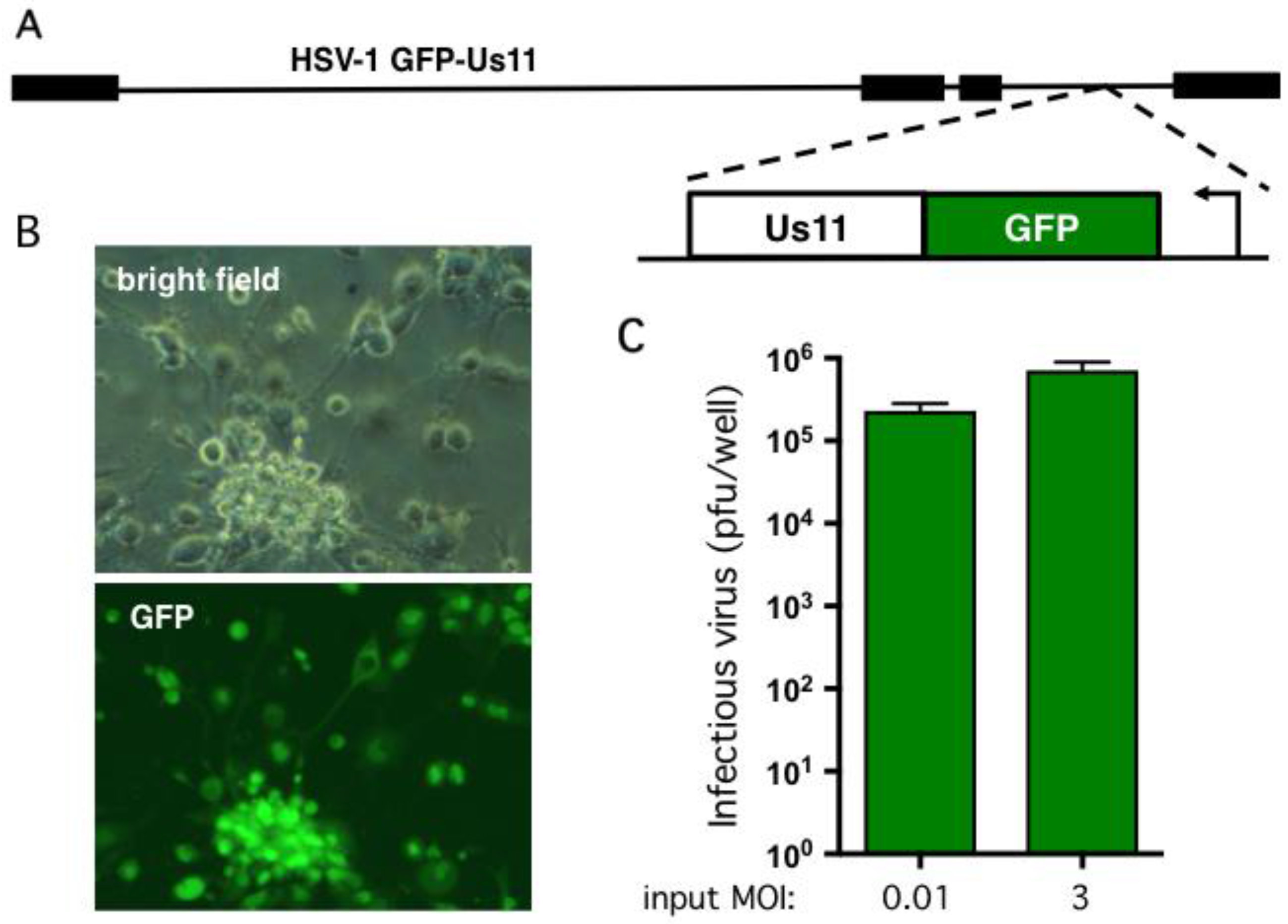
2.2. Human Neurons Are Permissive for HSV-1 Replication
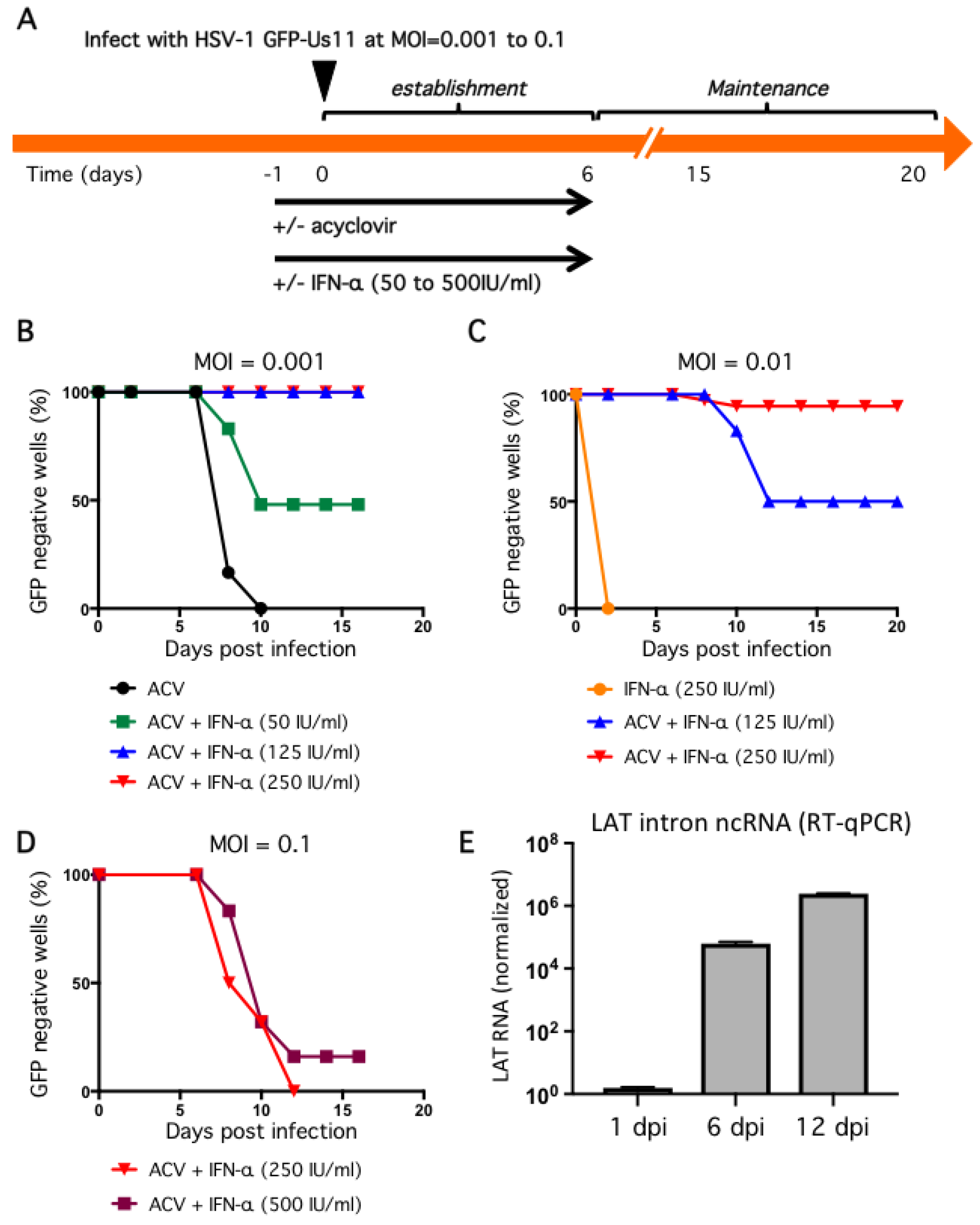
2.3. Non-Productive Infections Can Be Established Using a Combination of Low Inoculum, Acyclovir, and High Dose IFN-α
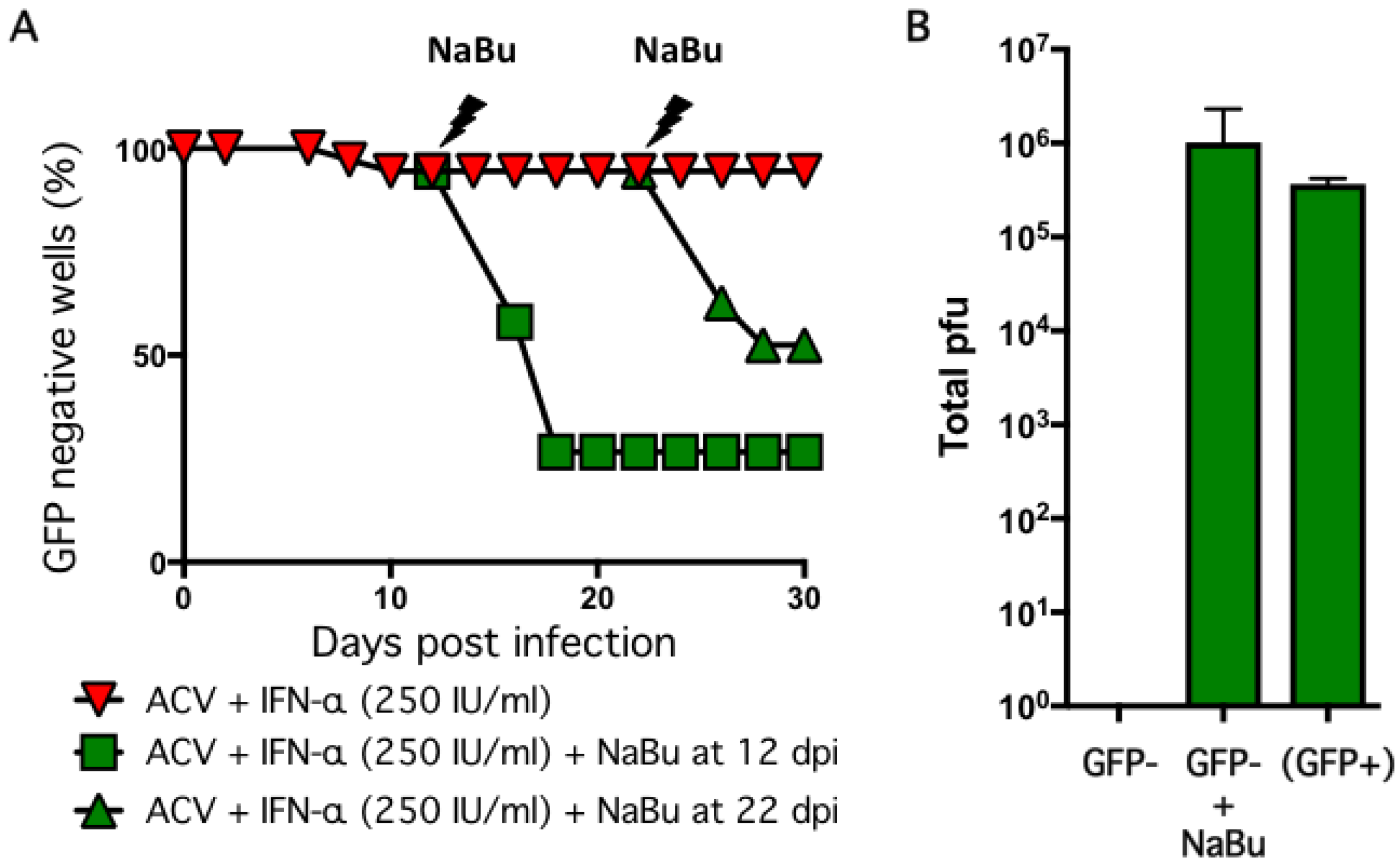
2.4. Non-Productive Infections Reinitiate Productive Replication upon Treatment with Sodium Butyrate
3. Discussion
4. Materials and Methods
4.1. Generation of Human Neural Stem Cells and Differentiated Neurons
4.2. Indirect Immunofluorescence Analysis
4.3. Infection of hESC-Derived Neurons with HSV-1
4.4. RNA Isolation and Transcript Analysis by Quantitative Reverse Transcription PCR (qRT-PCR)
- GAPDH FW: 5′-TCTTTTGCGTCGCCAGCCGA-3′
- GAPDH RV: 5′-ACCAGGCGCCCAATACGACC-3′
- LAT FW: 5′-AGTCCGGGCGGGCAGGCGCT-3′
- LAT RV: 5′-GCCCGGGCTGCCTGACCACCGAT-3′
4.5. Reactivation and Determination of Viral Titer
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Knipe, D.M.; Howley, P.M. Fields Virology, 6th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 1+2, pp. 1–2664. [Google Scholar]
- Nagel, M.A.; Choe, A.; Traktinskiy, I.; Gilden, D.H. Burning mouth syndrome due to herpes simplex virus type 1. BMJ Case Rep. 2015, 2015, bcr2015209488. [Google Scholar] [CrossRef] [PubMed]
- Gilden, D.H.; Mahalingam, R.; Cohrs, R.J.; Tyler, K.L. Herpesvirus infections of the nervous system. Nat. Clin. Pract. Neurol. 2007, 3, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.K.; Bloom, D.C. Experimental investigation of herpes simplex virus latency. Clin. Microbiol. Rev. 1997, 10, 419–443. [Google Scholar] [PubMed]
- Wilson, A.C.; Mohr, I. A cultured affair: HSV latency and reactivation in neurons. Trends Microbiol. 2012, 20, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Webre, J.M.; Hill, J.M.; Nolan, N.M.; Clement, C.; McFerrin, H.E.; Bhattacharjee, P.S.; Hsia, V.; Neumann, D.M.; Foster, T.P.; Lukiw, W.J.; et al. Rabbit and mouse models of HSV-1 latency, reactivation, and recurrent eye diseases. J. Biomed. Biotechnol. 2012, 2012, 612316. [Google Scholar] [CrossRef] [PubMed]
- Laibson, P.R.; Kibrick, S. Recurrence of herpes simplex virus in rabbit eyes: Results of a three-year study. Investig. Ophthalmol. 1969, 8, 346–350. [Google Scholar]
- Hill, J.M.; Dudley, J.B.; Shimomura, Y.; Kaufman, H.E. Quantitation and kinetics of induced HSV-1 ocular shedding. Curr. Eye Res. 1986, 5, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Speck, P.G.; Simmons, A. Divergent molecular pathways of productive and latent infection with a virulent strain of herpes simplex virus type 1. J. Virol. 1991, 65, 4001–4005. [Google Scholar] [PubMed]
- Deatly, A.M.; Spivack, J.G.; Lavi, E.; O’Boyle, D.R.; Fraser, N.W. Latent herpes simplex virus type 1 transcripts in peripheral and central nervous system tissues of mice map to similar regions of the viral genome. J. Virol. 1988, 62, 749–756. [Google Scholar] [PubMed]
- Spivack, J.G.; Fraser, N.W. Detection of herpes simplex virus type 1 transcripts during latent infection in mice. J. Virol. 1987, 61, 3841–3847. [Google Scholar] [PubMed]
- Devi-Rao, G.B.; Bloom, D.C.; Stevens, J.G.; Wagner, E.K. Herpes simplex virus type 1 DNA replication and gene expression during explant-induced reactivation of latently infected murine sensory ganglia. J. Virol. 1994, 68, 1271–1282. [Google Scholar] [PubMed]
- Knotts, F.B.; Cook, M.L.; Stevens, J.G. Pathogenesis of herpetic encephalitis in mice after ophthalmic inoculation. J. Infect. Dis. 1974, 130, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Leib, D.A.; Coen, D.M.; Bogard, C.L.; Hicks, K.A.; Yager, D.R.; Knipe, D.M.; Tyler, K.L.; Schaffer, P.A. Immediate-early regulatory gene mutants define different stages in the establishment and reactivation of herpes simplex virus latency. J. Virol. 1989, 63, 759–768. [Google Scholar] [PubMed]
- Sawtell, N.M.; Thompson, R.L. Rapid in vivo reactivation of herpes simplex virus in latently infected murine ganglionic neurons after transient hyperthermia. J. Virol. 1992, 66, 2150–2156. [Google Scholar] [PubMed]
- Laycock, K.A.; Lee, S.F.; Brady, R.H.; Pepose, J.S. Characterization of a murine model of recurrent herpes simplex viral keratitis induced by ultraviolet B radiation. Investig. Ophthalmol. Vis. Sci. 1991, 32, 2741–2746. [Google Scholar]
- Steiner, I.; Mador, N.; Reibstein, I.; Spivack, J.G.; Fraser, N.W. Herpes simplex virus type 1 gene expression and reactivation of latent infection in the central nervous system. Neuropathol. Appl. Neurobiol. 1994, 20, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Verweij, M.C.; Ressing, M.E.; Knetsch, W.; Quinten, E.; Halenius, A.; van Bel, N.; Hengel, H.; Drijfhout, J.W.; van Hall, T.; Wiertz, E.J.H.J. Inhibition of mouse TAP by immune evasion molecules encoded by non-murine herpesviruses. Mol. Immunol. 2011, 48, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Mandarino, A.; Chao, M.V.; Mohr, I.; Wilson, A.C. Transient Reversal of Episome Silencing Precedes VP16-Dependent Transcription during Reactivation of Latent HSV-1 in Neurons. PLoS Pathog. 2012, 8, e1002540. [Google Scholar] [CrossRef] [PubMed]
- Cleary, M.A.; Stern, S.; Tanaka, M.; Herr, W. Differential positive control by Oct-1 and Oct-2: Activation of a transcriptionally silent motif through Oct-1 and VP16 corecruitment. Genes Dev. 1993, 7, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Camarena, V.; Kobayashi, M.; Kim, J.Y.; Roehm, P.C.; Perez, R.; Gardner, J.; Wilson, A.C.; Mohr, I.; Chao, M.V. Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV-1 latency in neurons. Cell Host Microbe 2010, 8, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Wilson, A.C.; Chao, M.V.; Mohr, I. Control of viral latency in neurons by axonal mTOR signaling and the 4E-BP translation repressor. Genes Dev. 2012, 26, 1527–1532. [Google Scholar] [CrossRef] [PubMed]
- Cliffe, A.R.; Arbuckle, J.H.; Vogel, J.L.; Geden, M.J.; Rothbart, S.B.; Cusack, C.L.; Strahl, B.D.; Kristie, T.M.; Deshmukh, M. Neuronal Stress Pathway Mediating a Histone Methyl/Phospho Switch Is Required for Herpes Simplex Virus Reactivation. Cell Host Microbe 2015, 18, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Jurak, I.; Hackenberg, M.; Kim, J.Y.; Pesola, J.M.; Everett, R.D.; Preston, C.M.; Wilson, A.C.; Coen, D.M. Expression of Herpes Simplex Virus 1 miRNAs in Cell Culture Models of Quiescent and Latent Infection. J. Virol. 2014, 88, 2337–2339. [Google Scholar] [CrossRef] [PubMed]
- Linderman, J.A.; Kobayashi, M.; Rayannavar, V.; Fak, J.J.; Darnell, R.B.; Chao, M.V.; Wilson, A.C.; Mohr, I. Immune Escape via a Transient Gene Expression Program Enables Productive Replication of a Latent Pathogen. Cell Rep. 2017, 18, 1312–1323. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Shiflett, L.A.; Linderman, J.A.; Mohr, I.; Wilson, A.C. Using homogeneous primary neuron cultures to study fundamental aspects of HSV-1 latency and reactivation. Methods Mol. Biol. 2014, 1144, 167–179. [Google Scholar] [PubMed]
- Kobayashi, M.; Kim, J.Y.; Camarena, V.; Roehm, P.C.; Chao, M.V.; Wilson, A.C.; Mohr, I. A primary neuron culture system for the study of herpes simplex virus latency and reactivation. J. Vis. Exp. 2012, 62, e3823. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, C.L.; Smith, R.L.; Freed, C.R.; Johnson, E.M. Nerve growth factor-dependence of herpes simplex virus latency in peripheral sympathetic and sensory neurons in vitro. J. Neurosci. 1990, 10, 1268–1275. [Google Scholar] [PubMed]
- Wilcox, C.L.; Johnson, E.M. Characterization of nerve growth factor-dependent herpes simplex virus latency in neurons in vitro. J. Virol. 1988, 62, 393–399. [Google Scholar] [PubMed]
- Wilcox, C.L.; Johnson, E.M. Nerve growth factor deprivation results in the reactivation of latent herpes simplex virus in vitro. J. Virol. 1987, 61, 2311–2315. [Google Scholar] [PubMed]
- Roehm, P.C.; Camarena, V.; Nayak, S.; Gardner, J.B.; Wilson, A.; Mohr, I.; Chao, M.V. Cultured vestibular ganglion neurons demonstrate latent HSV1 reactivation. Laryngoscope 2011, 121, 2268–2275. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.A.; Nayak, S.; Camarena, V.; Gardner, J.; Wilson, A.; Mohr, I.; Chao, M.V.; Roehm, P.C. A cell culture model of facial palsy resulting from reactivation of latent herpes simplex type 1. Otol. Neurotol. 2012, 33, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Avgousti, D.C.; Weitzman, M.D. Stress Flips a Chromatin Switch to Wake Up Latent Virus. Cell Host Microbe 2015, 18, 639–641. [Google Scholar] [CrossRef] [PubMed]
- Cliffe, A.R.; Wilson, A.C. Restarting Lytic Gene Transcription at the Onset of Herpes Simplex Virus Reactivation. J. Virol. 2017, 91, e01419-16. [Google Scholar] [CrossRef] [PubMed]
- Azarkh, Y.; Bos, N.; Gilden, D.H.; Cohrs, R.J. Human trigeminal ganglionic explants as a model to study alphaherpesvirus reactivation. J. Neurovirol. 2012, 18, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Cohrs, R.J.; Laguardia, J.J.; Gilden, D.H. Distribution of latent herpes simplex virus type-1 and varicella zoster virus DNA in human trigeminal Ganglia. Virus Genes 2005, 31, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Cohrs, R.J.; Randall, J.; Smith, J.; Gilden, D.H.; Dabrowski, C.; van Der Keyl, H.; Tal-Singer, R. Analysis of individual human trigeminal ganglia for latent herpes simplex virus type 1 and varicella-zoster virus nucleic acids using real-time PCR. J. Virol. 2000, 74, 11464–11471. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Seitz, S.; Pointon, T.; Bowlin, J.L.; Cohrs, R.J.; Jonjic, S.; Haas, J.; Wellish, M.; Gilden, D.H. Varicella zoster virus infection of highly pure terminally differentiated human neurons. J. Neurovirol. 2013, 19, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Pugazhenthi, S.; Nair, S.; Velmurugan, K.; Liang, Q.; Mahalingam, R.; Cohrs, R.J.; Nagel, M.A.; Gilden, D.H. Varicella-zoster virus infection of differentiated human neural stem cells. J. Virol. 2011, 85, 6678–6686. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Zhou, W.; Scott-McKean, J.J.; Emmerling, K.L.; Cai, G.-Y.; Krah, D.L.; Costa, A.C.; Freed, C.R.; Levin, M.J. Human sensory neurons derived from induced pluripotent stem cells support varicella-zoster virus infection. PLoS ONE 2012, 7, e53010. [Google Scholar] [CrossRef] [PubMed]
- Markus, A.; Grigoryan, S.; Sloutskin, A.; Yee, M.B.; Zhu, H.; Yang, I.H.; Thakor, N.V.; Sarid, R.; Kinchington, P.R.; Goldstein, R.S. Varicella-zoster virus (VZV) infection of neurons derived from human embryonic stem cells: Direct demonstration of axonal infection, transport of VZV, and productive neuronal infection. J. Virol. 2011, 85, 6220–6233. [Google Scholar] [CrossRef] [PubMed]
- Sadaoka, T.; Depledge, D.P.; Rajbhandari, L.; Venkatesan, A.; Breuer, J.; Cohen, J.I. In vitro system using human neurons demonstrates that varicella-zoster vaccine virus is impaired for reactivation, but not latency. Proc. Natl. Acad. Sci. USA 2016, 113, E2403–E2412. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed]
- Tabar, V.; Panagiotakos, G.; Greenberg, E.D.; Chan, B.K.; Sadelain, M.; Gutin, P.H.; Studer, L. Migration and differentiation of neural precursors derived from human embryonic stem cells in the rat brain. Nat. Biotechnol. 2005, 23, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Placantonakis, D.G.; Tomishima, M.J.; Lafaille, F.; Desbordes, S.C.; Jia, F.; Socci, N.D.; Viale, A.; Lee, H.; Harrison, N.; Tabar, V.; et al. BAC transgenesis in human embryonic stem cells as a novel tool to define the human neural lineage. Stem Cells 2009, 27, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Edri, R.; Yaffe, Y.; Ziller, M.J.; Mutukula, N.; Volkman, R.; David, E.; Jacob-Hirsch, J.; Malcov, H.; Levy, C.; Rechavi, G.; et al. Analysing human neural stem cell ontogeny by consecutive isolation of Notch active neural progenitors. Nat. Commun. 2015, 6, 6500. [Google Scholar] [CrossRef] [PubMed]
- Lendahl, U.; Zimmerman, L.B.; McKay, R.D. CNS stem cells express a new class of intermediate filament protein. Cell 1990, 60, 585–595. [Google Scholar] [CrossRef]
- Dahlstrand, J.; Lardelli, M.; Lendahl, U. Nestin mRNA expression correlates with the central nervous system progenitor cell state in many, but not all, regions of developing central nervous system. Brain Res. Dev. Brain Res. 1995, 84, 109–129. [Google Scholar] [CrossRef]
- D’Aiuto, L.; Zhi, Y.; Kumar Das, D.; Wilcox, M.R.; Johnson, J.W.; McClain, L.; MacDonald, M.L.; Di Maio, R.; Schurdak, M.E.; Piazza, P.; et al. Large-scale generation of human iPSC-derived neural stem cells/early neural progenitor cells and their neuronal differentiation. Organogenesis 2014, 10, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.P.; Couillard-Després, S.; Cooper-Kuhn, C.M.; Winkler, J.; Aigner, L.; Kuhn, H.G. Transient expression of doublecortin during adult neurogenesis. J. Comp. Neurol. 2003, 467, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Walker, T.L.; Yasuda, T.; Adams, D.J.; Bartlett, P.F. The doublecortin-expressing population in the developing and adult brain contains multipotential precursors in addition to neuronal-lineage cells. J. Neurosci. 2007, 27, 3734–3742. [Google Scholar] [CrossRef] [PubMed]
- Benboudjema, L.; Mulvey, M.; Gao, Y.; Pimplikar, S.W.; Mohr, I. Association of the herpes simplex virus type 1 Us11 gene product with the cellular kinesin light-chain-related protein PAT1 results in the redistribution of both polypeptides. J. Virol. 2003, 77, 9192–9203. [Google Scholar] [CrossRef] [PubMed]
- Phelan, D.; Barrozo, E.R.; Bloom, D.C. HSV1 latent transcription and non-coding RNA: A critical retrospective. J. Neuroimmunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Galloway, D.A.; Fenoglio, C.; Shevchuk, M.; McDougall, J.K. Detection of herpes simplex RNA in human sensory ganglia. Virology 1979, 95, 265–268. [Google Scholar] [CrossRef]
- Hafezi, W.; Lorentzen, E.U.; Eing, B.R.; Müller, M.; King, N.J. C.; Klupp, B.; Mettenleiter, T.C.; Kühn, J.E. Entry of Herpes Simplex Virus Type 1 (HSV-1) into the Distal Axons of Trigeminal Neurons Favors the Onset of Nonproductive, Silent Infection. PLoS Pathog. 2012, 8, e1002679. [Google Scholar] [CrossRef] [PubMed]
- Daigle, D.; Gradoville, L.; Tuck, D.; Schulz, V.; Wang’ondu, R.; Ye, J.; Gorres, K.; Miller, G. Valproic acid antagonizes the capacity of other histone deacetylase inhibitors to activate the Epstein-barr virus lytic cycle. J. Virol. 2011, 85, 5628–5643. [Google Scholar] [CrossRef] [PubMed]
- Luka, J.; Kallin, B.; Klein, G. Induction of the Epstein-Barr virus (EBV) cycle in latently infected cells by n-butyrate. Virology 1979, 94, 228–231. [Google Scholar] [CrossRef]
- Ertel, M.K.; Cammarata, A.L.; Hron, R.J.; Neumann, D.M. CTCF occupation of the HSV-1 genome is disrupted at early times post-reactivation in a transcription-dependent manner. J. Virol. 2012, 86, 12741–12759. [Google Scholar] [CrossRef] [PubMed]
- Danaher, R.J.; Jacob, R.J.; Steiner, M.R.; Allen, W.R.; Hill, J.M.; Miller, C.S. Histone deacetylase inhibitors induce reactivation of herpes simplex virus type 1 in a latency-associated transcript-independent manner in neuronal cells. J. Neurovirol. 2005, 11, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Neumann, D.M.; Bhattacharjee, P.S.; Hill, J.M. Sodium butyrate: A chemical inducer of in vivo reactivation of herpes simplex virus type 1 in the ocular mouse model. J. Virol. 2007, 81, 6106–6110. [Google Scholar] [CrossRef] [PubMed]
- Yanez, A.; Harrell, T.; Sriranganathan, H.; Ives, A.; Bertke, A. Neurotrophic Factors NGF, GDNF and NTN Selectively Modulate HSV1 and HSV2 Lytic Infection and Reactivation in Primary Adult Sensory and Autonomic Neurons. Pathogens 2017, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Enquist, L.W.; Leib, D.A. Intrinsic and innate defenses of neurons: Détente with the herpesviruses. J. Virol. 2016, 91. [Google Scholar] [CrossRef] [PubMed]
- Leib, D.A.; Harrison, T.E.; Laslo, K.M.; Machalek, M.A.; Moorman, N.J.; Virgin, H.W. Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses in vivo. J. Exp. Med. 1999, 189, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Luker, G.D.; Prior, J.L.; Song, J.; Pica, C.M.; Leib, D.A. Bioluminescence imaging reveals systemic dissemination of herpes simplex virus type 1 in the absence of interferon receptors. J. Virol. 2003, 77, 11082–11093. [Google Scholar] [CrossRef] [PubMed]
- Rosato, P.C.; Leib, D.A. Neuronal Interferon Signaling Is Required for Protection against Herpes Simplex Virus Replication and Pathogenesis. PLoS Pathog. 2015, 11, e1005028. [Google Scholar] [CrossRef] [PubMed]
- Bloom, D.C. Alphaherpesvirus Latency: A Dynamic State of Transcription and Reactivation. Adv. Virus Res. 2016, 94, 53–80. [Google Scholar] [PubMed]
- Orzalli, M.H.; Conwell, S.E.; Berrios, C.; DeCaprio, J.A.; Knipe, D.M. Nuclear interferon-inducible protein 16 promotes silencing of herpesviral and transfected DNA. Proc. Natl. Acad. Sci. USA 2013, 110, E4492–E4501. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.I.; De Andrea, M.; Borgogna, C.; Albertini, S.; Landini, M.M.; Peretti, A.; Johnson, K.E.; Chandran, B.; Landolfo, S.; Gariglio, M. The Nuclear DNA Sensor IFI16 Acts as a Restriction Factor for Human Papillomavirus Replication through Epigenetic Modifications of the Viral Promoters. J. Virol. 2015, 89, 7506–7520. [Google Scholar]
- Johnson, K.E.; Bottero, V.; Flaherty, S.; Dutta, S.; Singh, V.V.; Chandran, B. IFI16 Restricts HSV-1 Replication by Accumulating on the HSV-1 Genome, Repressing HSV-1 Gene Expression, and Directly or Indirectly Modulating Histone Modifications. PLoS Pathog. 2014, 10, e1004503. [Google Scholar] [CrossRef] [PubMed]
- Szpara, M.L.; Kobiler, O.; Enquist, L.W. A common neuronal response to alphaherpesvirus infection. J. Neuroimmune Pharmacol. 2010, 5, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Bertke, A.S.; Swanson, S.M.; Chen, J.; Imai, Y.; Kinchington, P.R.; Margolis, T.P. A5-positive primary sensory neurons are nonpermissive for productive infection with herpes simplex virus 1 in vitro. J. Virol. 2011, 85, 6669–6677. [Google Scholar] [CrossRef] [PubMed]
- Warren, K.G.; Brown, S.M.; Wroblewska, Z.; Gilden, D.H.; Koprowski, H.; Subak-Sharpe, J. Isolation of latent herpes simplex virus from the superior cervical and vagus ganglions of human beings. N. Engl. J. Med. 1978, 298, 1068–1069. [Google Scholar] [CrossRef] [PubMed]
- Lafferty, W.E.; Coombs, R.W.; Benedetti, J.; Critchlow, C.; Corey, L. Recurrences after oral and genital herpes simplex virus infection. Influence of site of infection and viral type. N. Engl. J. Med. 1987, 316, 1444–1449. [Google Scholar] [CrossRef] [PubMed]
- Mertens, J.; Marchetto, M.C.; Bardy, C.; Gage, F.H. Evaluating cell reprogramming, differentiation and conversion technologies in neuroscience. Nat. Rev. Neurosci. 2016, 17, 424–437. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, J.W.; Eade, K.T.; Szűcs, A.; Lo Sardo, V.; Tsunemoto, R.K.; Williams, D.; Sanna, P.P.; Baldwin, K.K. Selective conversion of fibroblasts into peripheral sensory neurons. Nat. Neurosci. 2015, 18, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Markus, A.; Lebenthal-Loinger, I.; Yang, I.H.; Kinchington, P.R.; Goldstein, R.S. An in vitro model of latency and reactivation of varicella zoster virus in human stem cell-derived neurons. PLoS Pathog. 2015, 11, e1004885. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.; Steain, M.; Slobedman, B.; Abendroth, A. Differentiated neuroblastoma cells provide a highly efficient model for studies of productive varicella-zoster virus infection of neuronal cells. J. Virol. 2011, 85, 8436–8442. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Yang, H.; Shi, Y.; Wei, M.; Xian, J.; Hu, W. [Establishment of a cell model system of herpes simplex virus type II latent infection and reactivation in SH-SY5Y cells]. Wei Sheng Wu Xue Bao 2010, 50, 98–106. [Google Scholar] [PubMed]
- Thellman, N.M.; Botting, C.; Madaj, Z.; Triezenberg, S.J. An Immortalized Human Dorsal Root Ganglia Cell Line Provides a Novel Context to Study Herpes Simplex Virus Type-1 Latency and Reactivation. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pourchet, A.; Modrek, A.S.; Placantonakis, D.G.; Mohr, I.; Wilson, A.C. Modeling HSV-1 Latency in Human Embryonic Stem Cell-Derived Neurons. Pathogens 2017, 6, 24. https://doi.org/10.3390/pathogens6020024
Pourchet A, Modrek AS, Placantonakis DG, Mohr I, Wilson AC. Modeling HSV-1 Latency in Human Embryonic Stem Cell-Derived Neurons. Pathogens. 2017; 6(2):24. https://doi.org/10.3390/pathogens6020024
Chicago/Turabian StylePourchet, Aldo, Aram S. Modrek, Dimitris G. Placantonakis, Ian Mohr, and Angus C. Wilson. 2017. "Modeling HSV-1 Latency in Human Embryonic Stem Cell-Derived Neurons" Pathogens 6, no. 2: 24. https://doi.org/10.3390/pathogens6020024
APA StylePourchet, A., Modrek, A. S., Placantonakis, D. G., Mohr, I., & Wilson, A. C. (2017). Modeling HSV-1 Latency in Human Embryonic Stem Cell-Derived Neurons. Pathogens, 6(2), 24. https://doi.org/10.3390/pathogens6020024