
From Delta to Omicron—Genetic Epidemiology of SARS-CoV-2 (hCoV-19) in Southern Poland
 , , , , and
, , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Group
2.2. SARS-CoV-2 Whole-Genome Sequencing (WGS)
2.3. Phylogenetic Analyses
2.4. Epidemiological Data Analysis
3. Results
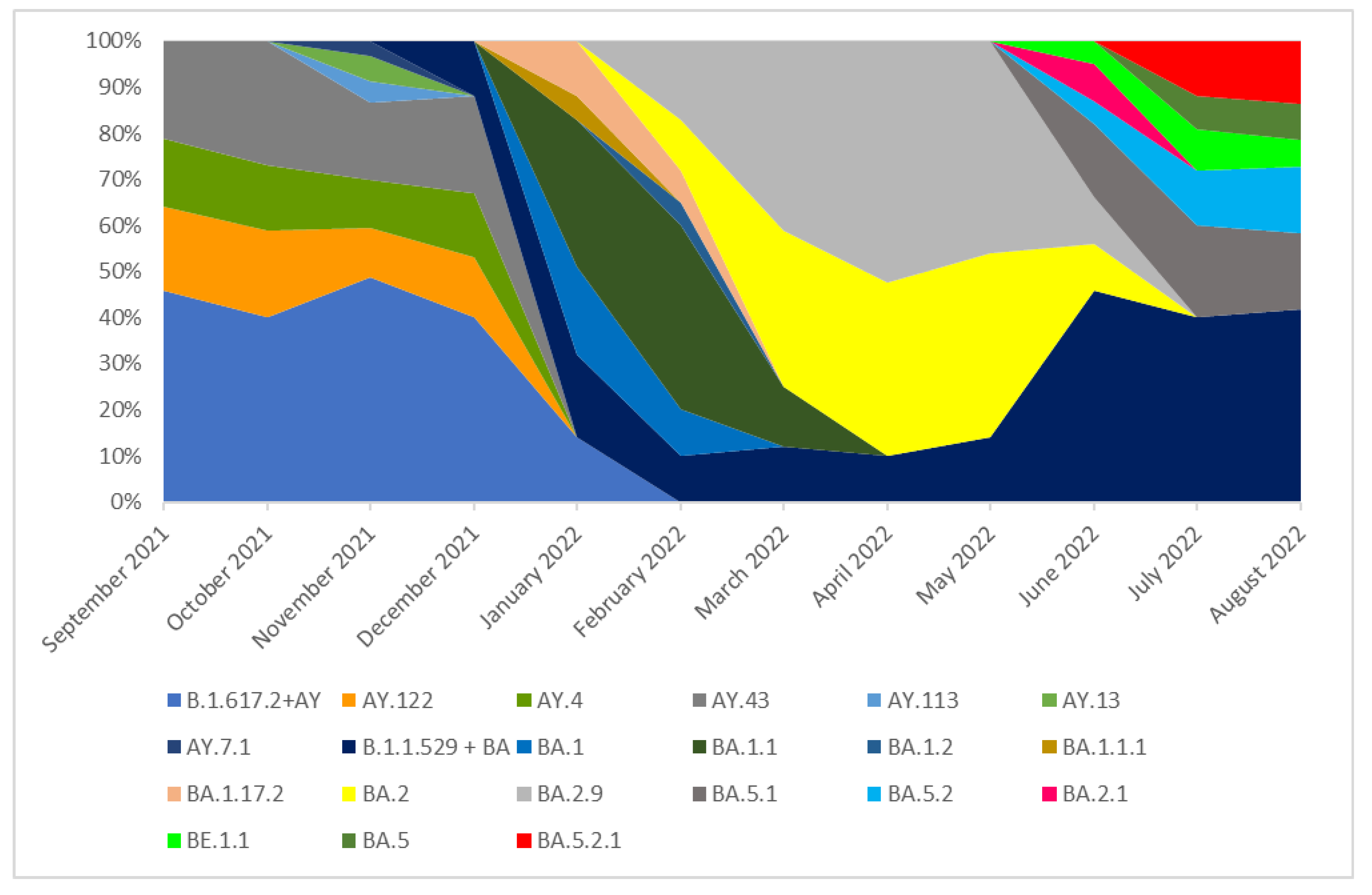
3.1. The Prevalence of SARS-CoV-2 Variants (hCoV-19) in Southern Poland
3.2. The Prevalence of SARS-CoV-2 Main Variants (hCoV-19) in Poland, the Czech Republic, and Slovakia
3.3. The Phylogenetic Connections of SARS-CoV-2 Clades
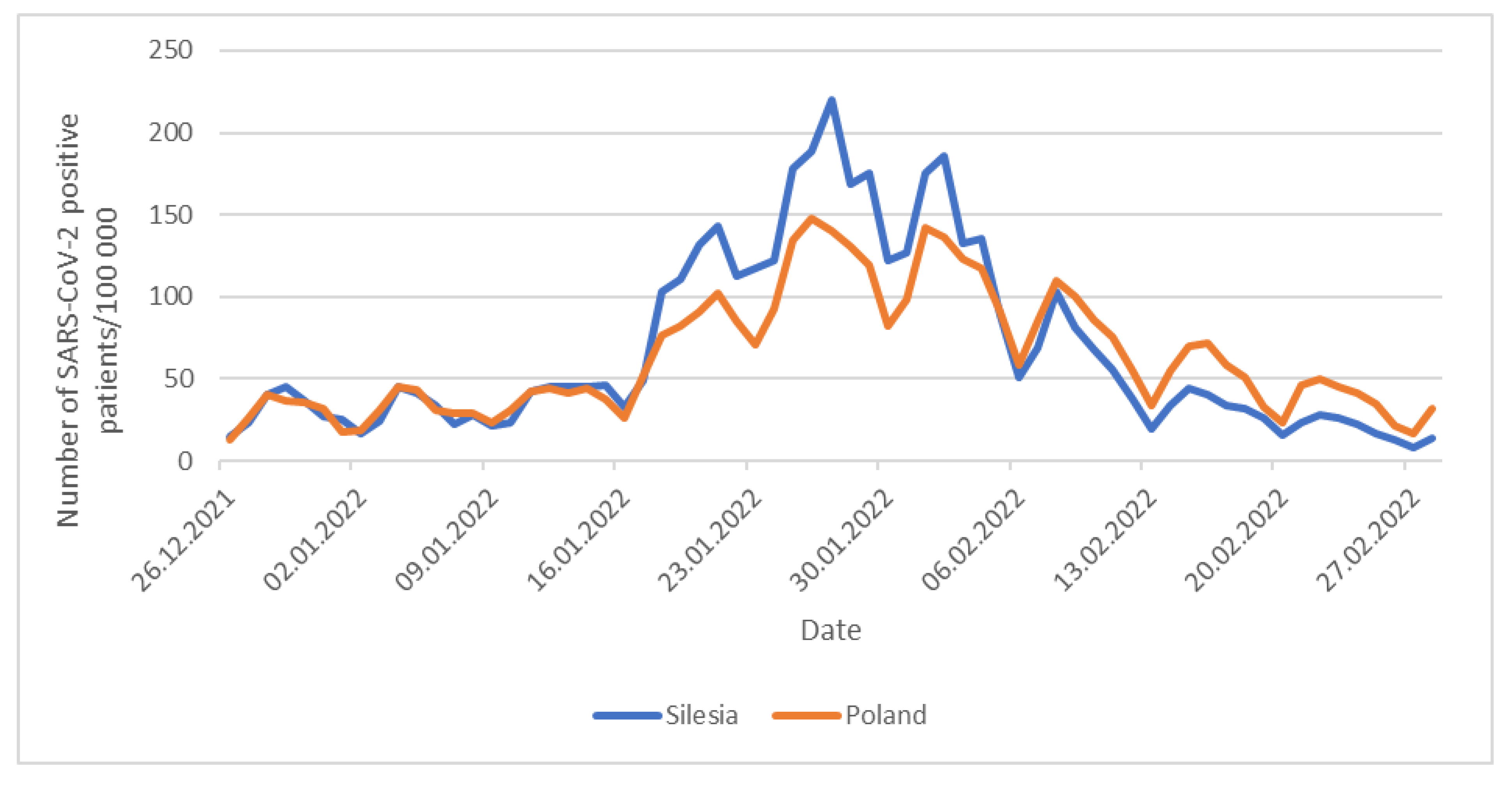
3.4. COVID-19 Cases in Silesia Compared to the Rest of Poland and Neighboring Countries
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Serwin, K.; Aksak-Wąs, B.; Parczewski, M. Phylodynamic Dispersal of SARS-CoV-2 Lineages Circulating across Polish–German Border Provinces. Viruses 2022, 14, 884. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Worobey, M.; Pekar, J.; Larsen, B.B.; Nelson, M.I.; Hill, V.; Joy, J.B.; Rambaut, A.; Suchard, M.A.; Wertheim, J.O.; Lemey, P. The emergence of SARS-CoV-2 in Europe and North America. Science 2020, 370, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. NextStrain: Real-Time Tracking of Pathogen Evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Munnink, B.B.O.; Worp, N.; Nieuwenhuijse, D.F.; Sikkema, R.S.; Haagmans, B.; Fouchier, R.A.M.; Koopmans, M. The next phase of SARS-CoV-2 Surveillance: Real-Time Molecular Epidemiology. Nat. Med. 2021, 27, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Cairo, A.; Iorio, M.V.; Spena, S.; Tagliabue, E.; Peyvandi, F.; Khudyakov, Y.E. Worldwide SARS-CoV-2 Haplotype Distribution in Early Pandemic. PLoS ONE 2022, 17, e0263705. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, M.I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. BBA-Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Thambiraja, T.S.; Karuppanan, K.; Subramaniam, G. Omicron and Delta Variant of SARS-CoV-2: A Comparative Computational Study of Spike Protein. J. Med. Virol. 2021, 94, 1641–1649. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.H.; Pires, D.E.; Ascher, D.B. DynaMut: Predicting the Impact of Mutations on Protein Conformation, Flexibility and Stability. Nucleic Acids Res. 2018, 46, W350–W355. [Google Scholar] [CrossRef] [PubMed]
- Laimer, J.; Hiebl-Flach, J.; Lengauer, D.; Lackner, P. MAESTROweb: A Web Server for Structure-Based Protein Stability Prediction. Bioinformatics 2016, 32, 1414–1416. [Google Scholar] [CrossRef] [PubMed]
- Pandurangan, A.P.; Ochoa-Montaño, B.; Ascher, D.B.; Blundell, T.L. SDM: A Server for Predicting Effects of Mutations on Protein Stability. Nucleic Acids Res. 2017, 45, W229–W235. [Google Scholar] [CrossRef] [PubMed]
- Abbasian, M.H.; Mahmanzar, M.; Rahimian, K.; Mahdavi, B.; Tokhanbigli, S.; Moradi, B.; Sisakht, M.M.; Deng, Y. Global Landscape of SARS-CoV-2 Mutations and Conserved Regions. J. Transl. Med. 2023, 21, 152. [Google Scholar] [CrossRef] [PubMed]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike Mutation D614G Alters SARS-CoV-2 Fitness. Nature 2021, 592, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Thao, T.T.N.; Hoffmann, D.; Taddeo, A.; Ebert, N.; Labroussaa, F.; Pohlmann, A.; King, J.; Steiner, S.; Kelly, J.N.; et al. SARS-CoV-2 Spike D614G Change Enhances Replication and Transmission. Nature 2021, 592, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, C.; Bhattacharya, M.; Sharma, A.R.; Mallik, B. Omicron (B.1.1.529)—A New Heavily Mutated Variant: Mapped Location and Probable Properties of Its Mutations with an Emphasis on S-Glycoprotein. Int. J. Biol. Macromol. 2022, 219, 980–997. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Zhang, X.; Gong, T.; Ma, J.; Zhang, P.; Cai, Z.; Ren, D.; Zhang, C. A Systematic Mutation Analysis of 13 Major SARS-CoV-2 Variants. Virus Res. 2024, 345, 199392. [Google Scholar] [CrossRef] [PubMed]
- Morawiec, E.; Miklasińska-Majdanik, M.; Bratosiewicz-Wąsik, J.; Wojtyczka, R.D.; Swolana, D.; Stolarek, I.; Czerwiński, M.; Skubis-Sikora, A.; Samul, M.; Polak, A.; et al. From Alpha to Delta—Genetic Epidemiology of SARS-CoV-2 (hCoV-19) in Southern Poland. Pathogens 2022, 11, 780. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://virological.org/t/masking-strategies-for-sars-cov-2-alignments/480 (accessed on 13 February 2025).
- Minh, B.Q.; Nguyen, M.A.T.; Von Haeseler, A. Ultrafast Approximation for Phylogenetic Bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-Likelihood Phylodynamic Analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.gov.pl/web/zdrowie/komunikat-ministra-zdrowia-w-zwiazku-z-dopuszczeniem-od-dnia-28-stycznia-2022-r-mozliwosci-przeprowadzania-testow-antygenowych-w-kierunku-sars-cov-2-przez-laboratoriummobilny-punkt-pobran (accessed on 13 February 2025).
- Available online: https://www.gov.pl/web/psse-zdunska-wola/zniesienie-obowiazku-noszenia-maseczek-zniesienie-kwarantanny-i-izolacji-domowej (accessed on 13 February 2025).
- Available online: https://koronawirusunas.pl (accessed on 13 February 2025).
- Tegally, H.; Moir, M.; Everatt, J.; Giovanetti, M.; Scheepers, C.; Wilkinson, E.; Subramoney, K.; Makatini, Z.; Moyo, S.; Amoako, D.G.; et al. Emergence of SARS-CoV-2 Omicron Lineages BA.4 and BA.5 in South Africa. Nat. Med. 2022, 28, 1785–1790. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Guo, Y.; Iketani, S.; Nair, M.S.; Li, Z.; Mohri, H.; Wang, M.; Yu, J.; Bowen, A.D.; Chang, J.Y.; et al. Antibody Evasion by SARS-CoV-2 Omicron Subvariants BA.2.12.1, BA.4 and BA.5. Nature 2022, 608, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yisimayi, A.; Jian, F.; Song, W.; Xiao, T.; Wang, L.; Du, S.; Wang, J.; Li, Q.; Chen, X.; et al. BA.2.12.1, BA.4 and BA.5 Escape Antibodies Elicited by Omicron Infection. Nature 2022, 608, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Uraki, R.; Kiso, M.; Iida, S.; Imai, M.; Takashita, E.; Kuroda, M.; Halfmann, P.J.; Loeber, S.; Maemura, T.; Yamayoshi, S.; et al. Characterization and Antiviral Susceptibility of SARS-CoV-2 Omicron BA.2. Nature 2022, 607, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Kimura, I.; Yamasoba, D.; Tamura, T.; Nao, N.; Suzuki, T.; Oda, Y.; Mitoma, S.; Ito, J.; Nasser, H.; Zahradnik, J.; et al. Virological Characteristics of the SARS-CoV-2 Omicron BA.2 Subvariants, Including BA.4 and BA.5. Cell 2022, 185, 3992–4007. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Wong, L.-Y.R.; Arora, P.; Zhang, L.; Rocha, C.; Odle, A.; Nehlmeier, I.; Kempf, A.; Richter, A.; Halwe, N.J.; et al. Omicron Subvariant BA.5 Efficiently Infects Lung Cells. Nat. Commun. 2023, 14, 3500. [Google Scholar] [CrossRef] [PubMed]
- Pastorio, C.; Noettger, S.; Nchioua, R.; Zech, F.; Sparrer, K.M.; Kirchhoff, F. Impact of Mutations Defining SARS-CoV-2 Omicron Subvariants BA.2.12.1 and BA.4/5 on Spike Function and Neutralization. iScience 2023, 26, 108299. [Google Scholar] [CrossRef] [PubMed]
- Tuekprakhon, A.; Nutalai, R.; Dijokaite-Guraliuc, A.; Zhou, D.; Ginn, H.M.; Selvaraj, M.; Liu, C.; Mentzer, A.J.; Supasa, P.; Duyvesteyn, H.M.; et al. Antibody Escape of SARS-CoV-2 Omicron BA.4 and BA.5 from Vaccine and BA.1 Serum. Cell 2022, 185, 2422–2433. [Google Scholar] [CrossRef] [PubMed]
- Parums, D.V. Editorial: World Health Organization (WHO) Variants of Concern Lineages Under Monitoring (VOC-LUM) in Response to the Global Spread of Lineages and Sublineages of Omicron, or B.1.1.529, SARS-CoV-2. Med. Sci. Monit. 2022, 28, e937676. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.S.; Toyoda, M.; Ode, H.; Barabona, G.; Hamana, H.; Kitamatsu, M.; Kishi, H.; Motozono, C.; Iwatani, Y.; Ueno, T.; et al. Dissecting Naturally Arising Amino Acid Substitutions at Position L452 of SARS-CoV-2 Spike. J. Virol. 2022, 96, e0116222. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lee, K.; Lee, H.; Park, J.W.; Cho, S.; Park, J.; Mun, J.; Park, S.; Lee, C.; Lee, J.; et al. Genomic Analysis and Tracking of SARS-CoV-2 Variants in Gwangju, South Korea, From 2020 to 2022. Influ. Other Respir. Viruses 2024, 18, e13350. [Google Scholar] [CrossRef] [PubMed]
- Telenti, A.; Hodcroft, E.B.; Robertson, D.L. The Evolution and Biology of SARS-CoV-2 Variants. Cold Spring Harb. Perspect. Med. 2022, 12, a041390. [Google Scholar] [CrossRef] [PubMed]
- Skuza, K.; Rutyna, P.; Krzowski, L.; Rabalski, L.; Lepionka, T. Surveillance of SARS-CoV-2 Genetic Variants in the Polish Armed Forces Using Whole Genome Sequencing Analysis. Int. J. Mol. Sci. 2023, 24, 14851. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Bhattacharya, M.; Nag, S.; Dhama, K.; Chakraborty, C. A Detailed Overview of SARS-CoV-2 Omicron: Its Sub-Variants, Mutations and Pathophysiology, Clinical Characteristics, Immunological Landscape, Immune Escape, and Therapies. Viruses 2023, 15, 167. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, H.; Chasapis, C.T.; Kelaidonis, K.; Ligielli, I.; Moore, G.J.; Gadanec, L.K.; Zulli, A.; Apostolopoulos, V.; Mavromoustakos, T.; Matsoukas, J.M. Understanding the Driving Forces That Trigger Mutations in SARS-CoV-2: Mutational Energetics and the Role of Arginine Blockers in COVID-19 Therapy. Viruses 2022, 14, 1029. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, H.; Ntallis, C.; Chasapis, C.T.; Kelaidonis, K.; Matsoukas, M.-T.; Plotas, P.; Apostolopoulos, V.; Moore, G.; Tsiodras, S.; Paraskevis, D.; et al. Molecular Epidemiology of SARS-CoV-2: The Dominant Role of Arginine in Mutations and Infectivity. Viruses 2023, 15, 309. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, H.; Moore, G.J.; Mavromoustakos, T.; Tsiodras, S.; Ligielli, I.; Kelaidonis, K.; Chasapis, C.T.; Gadanec, L.K.; Zulli, A.; Apostolopoulos, V.; et al. Discovery of a New Generation of Angiotensin Receptor Blocking Drugs: Receptor Mechanisms and in Silico Binding to Enzymes Relevant to SARS-CoV-2. Comput. Struct. Biotechnol. J. 2022, 20, 2091–2111. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miklasińska-Majdanik, M.; Morawiec, E.; Bratosiewicz-Wąsik, J.; Serwin, K.; Pudełko, A.; Czerwiński, M.; Bednarska-Czerwińska, A.; Parczewski, M.; Wąsik, T.J. From Delta to Omicron—Genetic Epidemiology of SARS-CoV-2 (hCoV-19) in Southern Poland. Pathogens 2025, 14, 708. https://doi.org/10.3390/pathogens14070708
Miklasińska-Majdanik M, Morawiec E, Bratosiewicz-Wąsik J, Serwin K, Pudełko A, Czerwiński M, Bednarska-Czerwińska A, Parczewski M, Wąsik TJ. From Delta to Omicron—Genetic Epidemiology of SARS-CoV-2 (hCoV-19) in Southern Poland. Pathogens. 2025; 14(7):708. https://doi.org/10.3390/pathogens14070708
Chicago/Turabian StyleMiklasińska-Majdanik, Maria, Emilia Morawiec, Jolanta Bratosiewicz-Wąsik, Karol Serwin, Adam Pudełko, Michał Czerwiński, Anna Bednarska-Czerwińska, Miłosz Parczewski, and Tomasz J. Wąsik. 2025. "From Delta to Omicron—Genetic Epidemiology of SARS-CoV-2 (hCoV-19) in Southern Poland" Pathogens 14, no. 7: 708. https://doi.org/10.3390/pathogens14070708
APA StyleMiklasińska-Majdanik, M., Morawiec, E., Bratosiewicz-Wąsik, J., Serwin, K., Pudełko, A., Czerwiński, M., Bednarska-Czerwińska, A., Parczewski, M., & Wąsik, T. J. (2025). From Delta to Omicron—Genetic Epidemiology of SARS-CoV-2 (hCoV-19) in Southern Poland. Pathogens, 14(7), 708. https://doi.org/10.3390/pathogens14070708