

Phylogeographic Analysis of Clade 2.3.4.4b H5N1 in Serbia Reveals Repeated Introductions and Spread Across the Balkans
 , , , , and
, , , , and 
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Sample Preparation
2.3. Extraction of Nucleic Acid and qRT-PCR
2.4. Isolation on MDCK Cell Line
2.5. Metagenomic Sequencing and Bioinformatic Analysis
2.6. Phylogenetic and Phylogeographic Analysis
3. Results
3.1. Virus Isolation and Sequencing
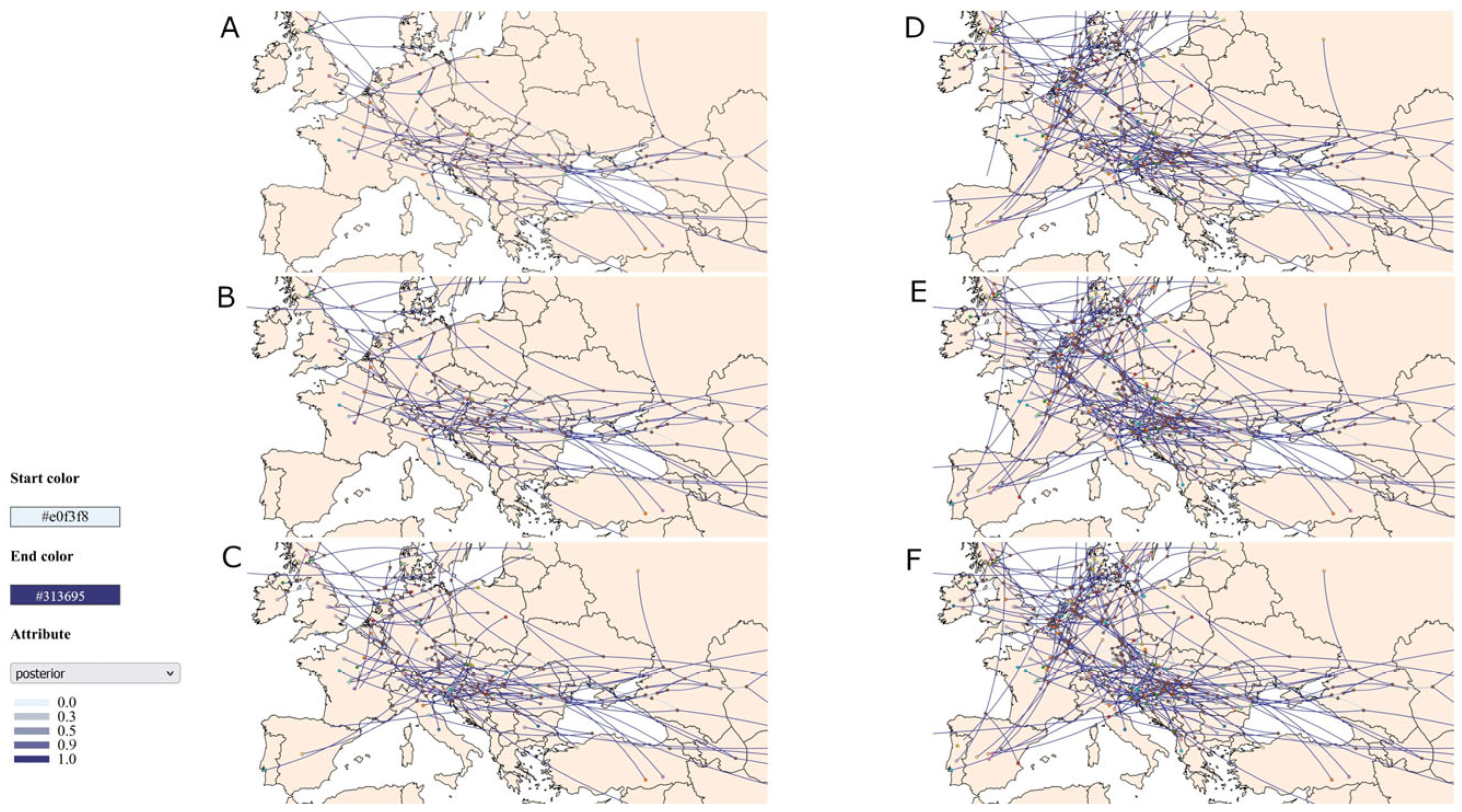
3.2. Results of Phylogenetic and Phylogeographic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Lambertucci, S.A.; Santangeli, A.; Plaza, P.I. The Threat of Avian Influenza H5N1 Looms over Global Biodiversity. Nat. Rev. Biodivers. 2025, 1, 7–9. [Google Scholar] [CrossRef]
- International Committee on Taxonomy of Viruses (ICTV). Virus Taxonomy: 2024 Relese; Master Species List (MSL #40); Alphainfluenzavirus Influenzae; ICTV: Bari, Italy, 2024. [Google Scholar]
- Peiris, J.S.M.; de Jong, M.D.; Guan, Y. Avian Influenza Virus (H5N1): A Threat to Human Health. Clin. Microbiol. Rev. 2007, 20, 243–267. [Google Scholar] [CrossRef] [PubMed]
- FAO. Global Consultation on Highly Pathogenic Avian Influenza (HPAI); FAO Animal Production and Health Reports, No. 20; FAO: Rome, Italy, 2023. [Google Scholar] [CrossRef]
- Wong, F.Y.; Yaqub, T.; Zhang, R.; Mukhtar, N.; Pervaiz, H.; Hussain Yawar, H.U.; Iqbal, M.; bin Aslam, H.; Aziz, M.W.; Akram, M.; et al. Highly Pathogenic Avian Influenza H5 Virus Exposure in Goats and Sheep. bioRxiv 2024. [Google Scholar] [CrossRef]
- World Organisation for Animal Health (WOAH). High Pathogenicity Avian Influenza (HPAI) in Cattle; WOAH: Brussels, Belgium, 2024. [Google Scholar]
- Smith, G.J.D.; Donis, R.O. Nomenclature Updates Resulting from the Evolution of Avian Influenza A(H5) Virus Clades 2.1.3.2a, 2.2.1, and 2.3.4 during 2013–2014. Influenza Other Respir. Viruses 2015, 9, 271–276. [Google Scholar] [CrossRef]
- Fusaro, A.; Zecchin, B.; Giussani, E.; Palumbo, E.; Agüero-García, M.; Bachofen, C.; Bálint, Á.; Banihashem, F.; Banyard, A.C.; Beerens, N.; et al. High Pathogenic Avian Influenza A(H5) Viruses of Clade 2.3.4.4b in Europe—Why Trends of Virus Evolution Are More Difficult to Predict. Virus Evol. 2024, 10, veae027. [Google Scholar] [CrossRef]
- Vaskovic, N.; Sekler, M.; Vidanovic, D.; Polacek, V.; Kukolj, V.; Matovic, K.; Jovanovic, M. Pathomorphological Lesions and Distribution of Viral Antigen in Birds Infected with the Pathogenic Strain of H5N1 Avian Influenza Virus. Acta Vet. Beogr. 2011, 61, 591–598. [Google Scholar] [CrossRef]
- Goletic, S.; Softic, A.; Omeragic, J.; Koro-Spahic, A.; Kapo, N.; Sabic, E.; Kasagic, D.; Goletic, T. Molecular Characterization and Phylogenetic Analysis of Highly Pathogenic H5N1 Clade 2.3.4.4b Virus in Bosnia and Herzegovina. Front. Vet. Sci. 2023, 10, 1255213. [Google Scholar] [CrossRef]
- Djurdjević, B.; Petrović, T.; Gajdov, V.; Vidanović, D.; Vučićević, I.; Samojlović, M.; Pajić, M. Natural Infection of Common Cranes (Grus grus) with Highly Pathogenic Avian Influenza H5N1 in Serbia. Front. Vet. Sci. 2024, 11, 1462546. [Google Scholar] [CrossRef]
- European Commission. Animal Diseases Reports 2024; European Commission: Brussels, Belgium, 2025. [Google Scholar]
- Statistical Office of Serbia. Farms by Size of Broiler Flock; Statistical Office of Serbia: Belgrade, Serbia, 2023. [Google Scholar]
- Shu, B.; Wu, K.-H.; Emery, S.; Villanueva, J.; Johnson, R.; Guthrie, E.; Berman, L.; Warnes, C.; Barnes, N.; Klimov, A.; et al. Design and Performance of the CDC Real-Time Reverse Transcriptase PCR Swine Flu Panel for Detection of 2009 A (H1N1) Pandemic Influenza Virus. J. Clin. Microbiol. 2011, 49, 2614–2619. [Google Scholar] [CrossRef]
- Chrzastek, K.; Lee, D.; Smith, D.; Sharma, P.; Suarez, D.L.; Pantin-Jackwood, M.; Kapczynski, D.R. Use of Sequence-Independent, Single-Primer-Amplification (SISPA) for Rapid Detection, Identification, and Characterization of Avian RNA Viruses. J. Viorol. 2017, 509, 159–166. [Google Scholar] [CrossRef]
- Šolaja, S.; Goletić, Š.; Veljović, L.; Glišić, D. Complex Patterns of WNV Evolution: A Focus on the Western Balkans and Central Europe. Front. Vet. Sci. 2024, 11, 1494746. [Google Scholar] [CrossRef] [PubMed]
- Aksamentov, I.; Roemer, C.; Hodcroft, E.; Neher, R. Nextclade: Clade Assignment, Mutation Calling and Quality Control for Viral Genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.; Yeo, W.; et al. GISAID’s Role in Pandemic Response. China CDC Wkly. 2021, 3, 1049–1051. [Google Scholar] [CrossRef] [PubMed]
- ESFLU WG2 Training School: Phylogenetics and Phylodynamics from Zero to Hero. Available online: http://zenodo.org/records/14234798 (accessed on 25 March 2025).
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A Fast Online Phylogenetic Tool for Maximum Likelihood Analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Welch, J.J.; Suchard, M.A. Phylogeography Takes a Relaxed Random Walk in Continuous Space and Time. Mol. Biol. Evol. 2010, 27, 1877–1885. [Google Scholar] [CrossRef]
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef]
- Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Mirinavičiūtė, G.; Niqueux, É.; Ståhl, K.; Staubach, C.; Terregino, C.; Willgert, K.; et al. Avian Influenza Overview September–December 2023. J. EFSA 2023, 21, e8539. [Google Scholar] [CrossRef]
- Alexakis, L.; Fusaro, A.; Kuiken, T.; Mirinavičiūtė, G.; Ståhl, K.; Staubach, C.; Svartström, O.; Terregino, C.; Willgert, K.; Delacourt, R.; et al. Avian Influenza Overview March–June 2024. J. EFSA 2024, 22, e8930. [Google Scholar] [CrossRef]
- Jindal, M.; Stone, H.; Lim, S.; MacIntyre, C.R. A Geospatial Perspective Toward the Role of Wild Bird Migrations and Global Poultry Trade in the Spread of Highly Pathogenic Avian Influenza H5N1. Geohealth 2025, 9, e2024GH001296. [Google Scholar] [CrossRef] [PubMed]
- Ojaste, I.; Leito, A.; Suorsa, P.; Hedenström, A.; Sepp, K.; Leivits, M.; Sellis, U.; Väli, Ü. From Northern Europe to Ethiopia: Long-Distance Migration of Common Cranes (Grus grus). Ornis Fenn. 2020, 97, 12–25. [Google Scholar] [CrossRef]
- Xu, Y.; Gong, P.; Wielstra, B.; Si, Y. Southward Autumn Migration of Waterfowl Facilitates Cross-Continental Transmission of the Highly Pathogenic Avian Influenza H5N1 Virus. Sci. Rep. 2016, 6, 30262. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šolaja, S.; Glišić, D.; Veljović, L.; Milošević, I.; Nićković, E.; Nišavić, J.; Milićević, V. Phylogeographic Analysis of Clade 2.3.4.4b H5N1 in Serbia Reveals Repeated Introductions and Spread Across the Balkans. Pathogens 2025, 14, 636. https://doi.org/10.3390/pathogens14070636
Šolaja S, Glišić D, Veljović L, Milošević I, Nićković E, Nišavić J, Milićević V. Phylogeographic Analysis of Clade 2.3.4.4b H5N1 in Serbia Reveals Repeated Introductions and Spread Across the Balkans. Pathogens. 2025; 14(7):636. https://doi.org/10.3390/pathogens14070636
Chicago/Turabian StyleŠolaja, Sofija, Dimitrije Glišić, Ljubiša Veljović, Ivan Milošević, Emilija Nićković, Jakov Nišavić, and Vesna Milićević. 2025. "Phylogeographic Analysis of Clade 2.3.4.4b H5N1 in Serbia Reveals Repeated Introductions and Spread Across the Balkans" Pathogens 14, no. 7: 636. https://doi.org/10.3390/pathogens14070636
APA StyleŠolaja, S., Glišić, D., Veljović, L., Milošević, I., Nićković, E., Nišavić, J., & Milićević, V. (2025). Phylogeographic Analysis of Clade 2.3.4.4b H5N1 in Serbia Reveals Repeated Introductions and Spread Across the Balkans. Pathogens, 14(7), 636. https://doi.org/10.3390/pathogens14070636