
Avian Influenza Virus: Comparative Evolution as the Key for Predicting Host Tropism Expansion


Abstract
1. Introduction
2. Materials and Methods
2.1. Identification of Proteins and Mutations Potentially Involved in Spillover
2.2. Protein Sequence Collection and Accession Number Identification
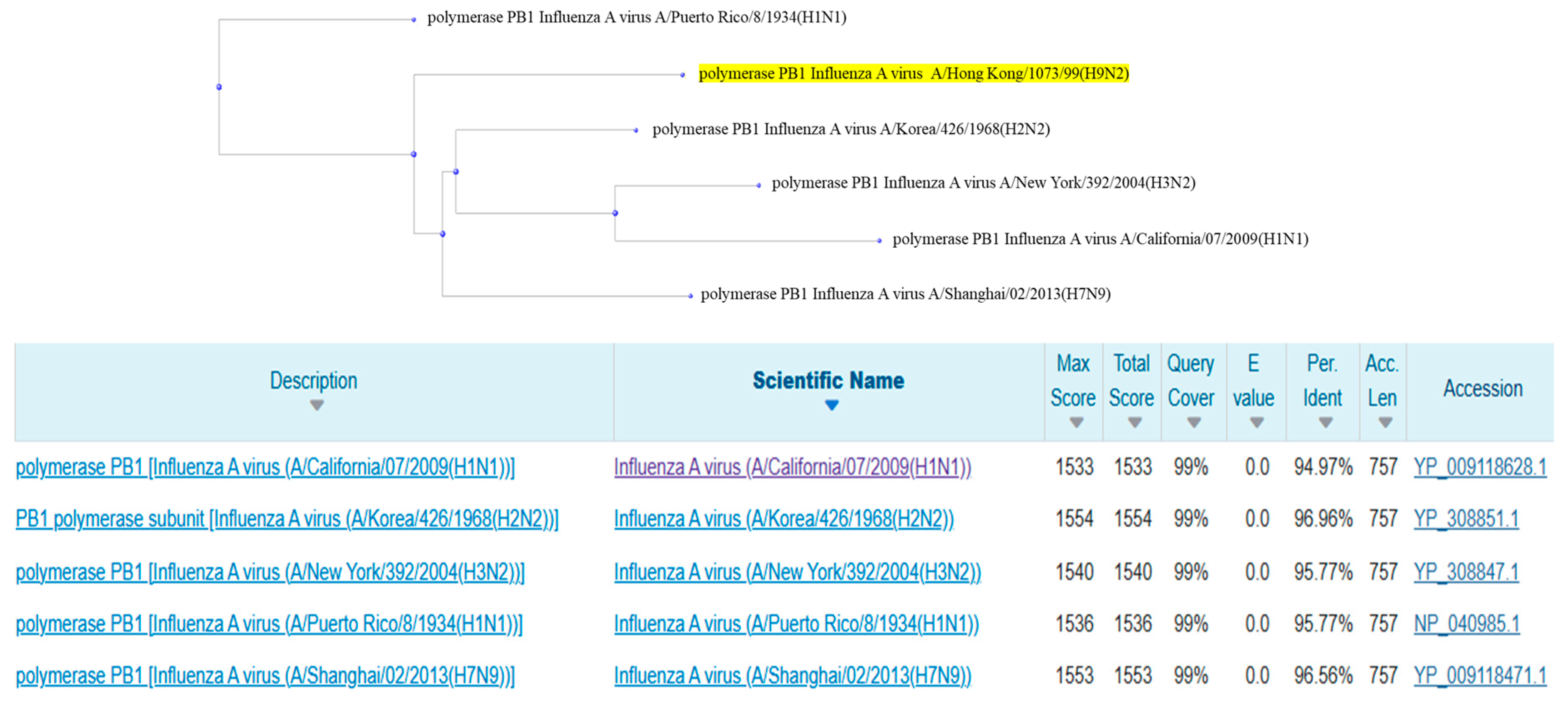
2.3. Alignment of Human Protein Sequences and Construction of Phylogenetic Trees
2.4. Analysis of Phylogenetic Trees and the Presence of Mutations in Aligned Sequences
3. Results
3.1. Proteins and Mutations Involved in Spillover
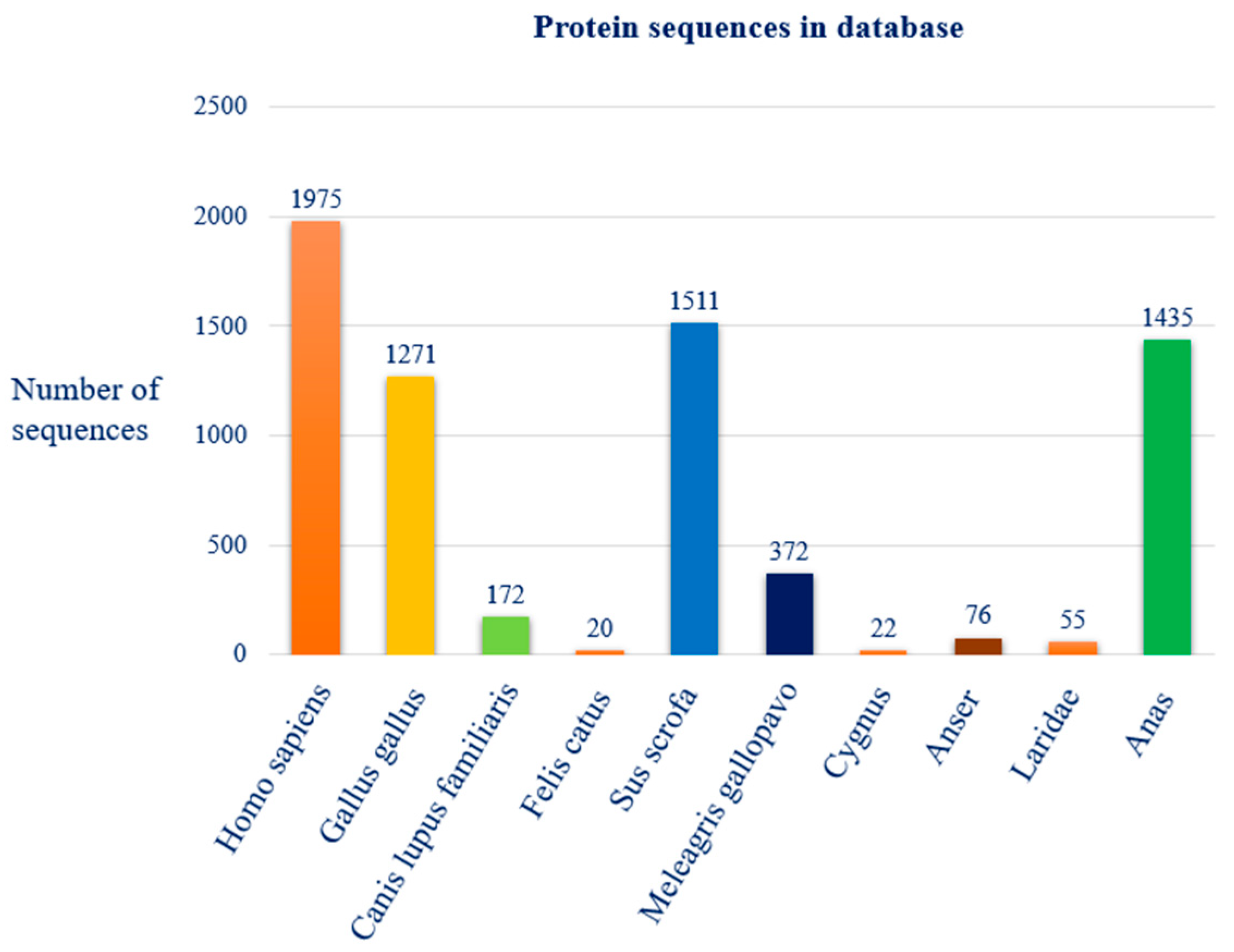
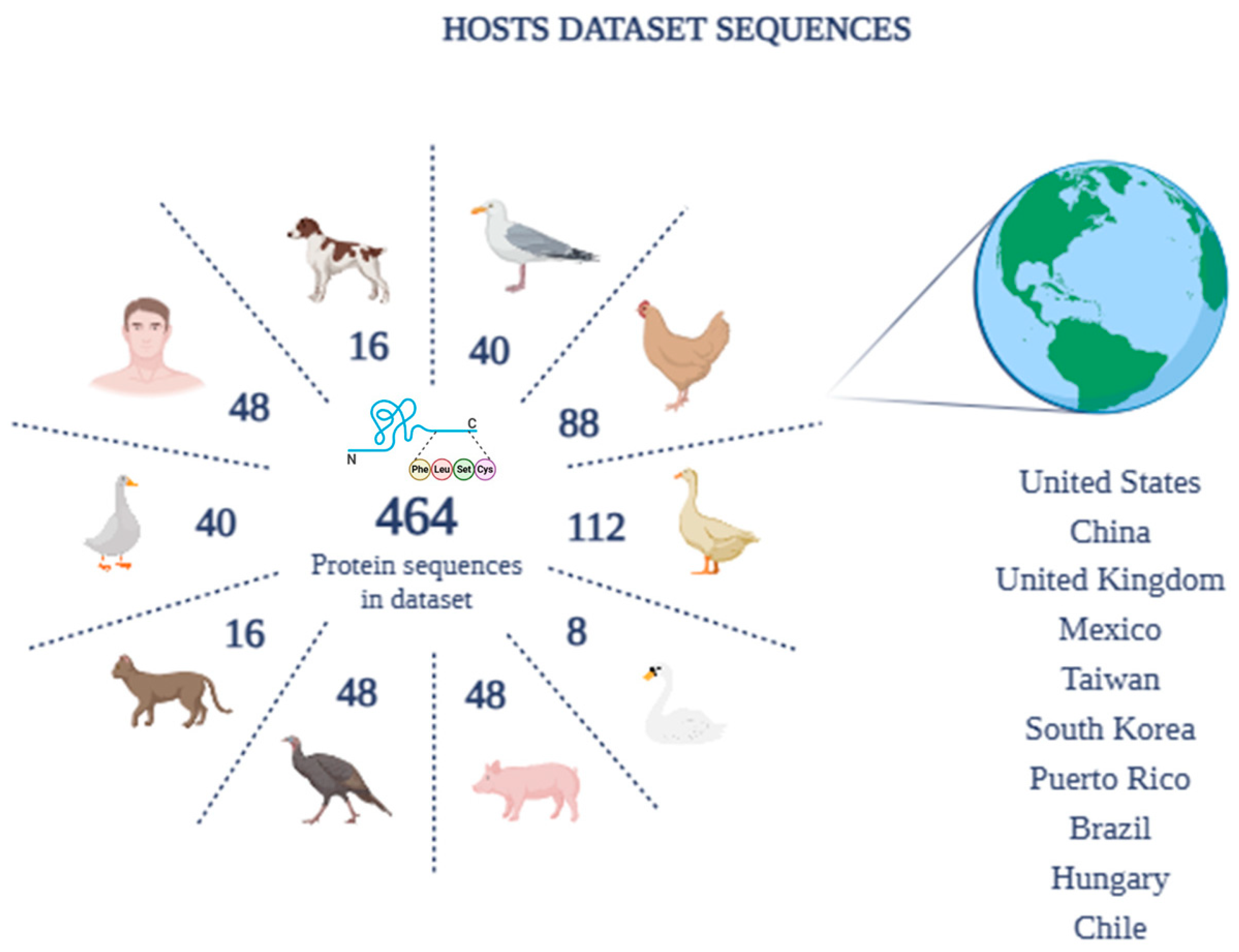
3.2. Protein Sequence Dataset
3.3. Alignment and Phylogenetic Relationship Analysis
3.4. Evolutionary Proximity and Shared Mutations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tilocca, B.; Paonessa, M.; De Salvo, M.; Roncada, P. Mitigating neglected zoonotic infections: A One Health approach on avian influenza in humans and animals. Gene Protein Dis. 2024, 3, 2327. [Google Scholar] [CrossRef]
- AbuBakar, U.; Amrani, L.; Kamarulzaman, F.A.; Karsani, S.A.; Hassandarvish, P.; Khairat, J.E. Avian Influenza Virus Tropism in Humans. Viruses 2023, 15, 833. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Kash, J.C.; Evolution, I.V. Host Adaptation and Pandemic Formation. Cell Host Microbe 2010, 7, 440. [Google Scholar] [CrossRef] [PubMed]
- Pavia, G.; Scarpa, F.; Ciccozzi, A.; Romano, C.; Branda, F.; Quirino, A.; Marascio, N.; Matera, G.; Sanna, D.; Ciccozzi, M. Changing and Evolution of Influenza Virus: Is It a Trivial Flu? Chemotherapy 2024, 69, 185–193. [Google Scholar] [CrossRef]
- Bisset, A.T.; Hoyne, G.F. Evolution and Adaptation of the Avian H7N9 Virus into the Human Host. Microorganism 2020, 8, 778. [Google Scholar] [CrossRef]
- Abdelwhab, E.M.; Mettenleiter, T.C. Zoonotic Animal Influenza Virus and Potential Mixing Vessel Hosts. Viruses 2023, 15, 980. [Google Scholar] [CrossRef]
- Guilligay, D.; Tarendeau, F.; Resa-Infante, P.; Coloma, R.; Crepin, T.; Sehr, P.; Lewis, J.; Ruigrok, R.W.H.; Ortin, J.; Hart, D.J.; et al. The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat. Struct. Mol. Biol. 2008, 15, 500–506. [Google Scholar] [CrossRef]
- Chakrabarti, A.K.; Pasricha, G. An insight into the PB1F2 protein and its multifunctional role in enhancing the pathogenicity of the influenza A viruses. Virology 2013, 440, 97–104. [Google Scholar] [CrossRef]
- Taubenberger, J.K.; Reid, A.H.; Lourens, R.M.; Wang, R.; Jin, G.; Fanning, T.G. Characterization of the 1918 influenza virus polymerase genes. Nature 2005, 437, 889–893. [Google Scholar] [CrossRef]
- Waters, K.; Wan, H.J.; Han, L.; Xue, J.; Ykema, M.; Tao, Y.J.; Wan, X.-F. Variations outside the conserved motifs of PB1 catalytic active site may affect replication efficiency of the RNP complex of influenza A virus. Virology 2021, 559, 145–155. [Google Scholar] [CrossRef]
- Lai, J.C.C.; Karunarathna, H.M.T.K.; Wong, H.H.; Peiris, J.S.M.; Nicholls, J.M. Neuraminidase activity and specificity of influenza A virus are influenced by haemagglutinin-receptor binding. Emerg. Microbes Infect. 2019, 8, 327–338. [Google Scholar] [CrossRef] [PubMed]
- An, S.-H.; Hong, S.-M.; Song, J.-H.; Son, S.-E.; Lee, C.-Y.; Choi, K.-S.; Kwon, H.-J. Engineering an Optimal Y280-Lineage H9N2 Vaccine Strain by Tuning PB2 Activity. Int. J. Mol. Sci. 2023, 24, 8840. [Google Scholar] [CrossRef] [PubMed]
- Voronina, O.; Ryzhova, N.; Aksenova, E.; Kunda, M.; Sharapova, N.; Fedyakina, I.; Chvala, I.; Borisevich, S.; Logunov, D.Y.; Gintsburg, A. Genetic features of highly pathogenic avian influenza viruses A(H5N8), isolated from the European part of the Russian Federation. Infect. Genet. Evol. 2018, 63, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Yeo, S.J.; Than, D.D.; Park, H.S.; Sung, H.W.; Park, H. Molecular Characterization of a Novel Avian Influenza A (H2N9) Strain Isolated from Wild Duck in Korea in 2018. Viruses 2019, 11, 1046. [Google Scholar] [CrossRef]
- Charostad, J.; Rukerd, M.R.Z.; Mahmoudvand, S.; Bashash, D.; Hashemi, S.M.A.; Nakhaie, M.; Zandi, K. A comprehensive review of highly pathogenic avian influenza (HPAI) H5N1: An imminent threat at doorstep. Travel Med. Infect. Dis. 2023, 55, 102638. [Google Scholar] [CrossRef]
- Qu, B.; Li, X.; Cardona, C.J.; Xing, Z. Reassortment and adaptive mutations of an emerging avian influenza virus H7N4 subtype in China. PLoS ONE 2020, 15, e0227597. [Google Scholar] [CrossRef]
- Yehia, N.; Rabie, N.; Adel, A.; Mossad, Z.; Nagshabandi, M.K.; Alharbi, M.T.; El-Saadony, M.T.; El-Tarabily, K.A.; Erfan, A. Differential replication characteristic of reassortant avian influenza A viruses H5N8 clade 2.3.4.4b in Madin-Darby canine kidney cell. Poult. Sci. 2023, 102, 102685. [Google Scholar] [CrossRef]
- Li, B.; Su, G.; Xiao, C.; Zhang, J.; Li, H.; Sun, N.; Lao, G.; Yu, Y.; Ren, X.; Qi, W.; et al. The PB2 co-adaptation of H10N8 avian influenza virus increases the pathogenicity to chickens and mice. Transbound. Emerg. Dis. 2022, 69, 1794–1803. [Google Scholar] [CrossRef]
- Li, X.; Jia, T.; Wang, K.; Wang, L.; Zhou, L.; Li, M.; Zhu, W.; Shu, Y.; Chen, Y. The PB2 I714S mutation influenced mammalian adaptation of the H3N2 canine influenza virus by interfering with nuclear import efficiency and RNP complex assembly. Emerg. Microbes Infect. 2024, 13, 2387439. [Google Scholar] [CrossRef]
- Guo, Y.; Bai, X.; Liu, Z.; Liang, B.; Zheng, Y.; Dankar, S.; Ping, J. Exploring the alternative virulence determinants PB2 S155N and PA S49Y/D347G that promote mammalian adaptation of the H9N2 avian influenza virus in mice. Vet. Res. 2023, 54, 97. [Google Scholar] [CrossRef]
- Ortiz, L.; Geiger, G.; Ferreri, L.; Moran, D.; Alvarez, D.; Gonzalez-Reiche, A.S.; Mendez, D.; Rajao, D.; Cordon-Rosales, C.; Perez, D.R.; et al. Evolution and Introductions of Influenza A Virus H1N1 in a Farrow-to-Finish Farm in Guatemala. Microbiol. Spectr. 2022, 11, e02878-22. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Chu, H.; Zhao, X.; Shuai, H.; Wong, B.H.-Y.; Wen, L.; Yuan, S.; Zheng, B.-J.; Zhou, J.; Yuen, K.-Y. Novel residues in the PA protein of avian influenza H7N7 virus affect virulence in mammalian hosts. Virology 2016, 498, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rolling, T.; Koerner, I.; Zimmermann, P.; Holz, K.; Haller, O.; Staeheli, P.; Kochs, G. Adaptive mutations resulting in enhanced polymerase activity contribute to high virulence of influenza A virus in mice. J. Virol. 2009, 83, 6673–6680. [Google Scholar] [CrossRef]
- Arai, Y.; Kawashita, N.; Elgendy, E.M.; Ibrahim, M.S.; Daidoji, T.; Ono, T.; Takagi, T.; Nakaya, T.; Matsumoto, K.; Watanabe, Y.; et al. PA Mutations Inherited during Viral Evolution Act Cooperatively To Increase Replication of Contemporary H5N1 Influenza Virus with an Expanded Host Range. J. Virol. 2020, 95, 10-1128. [Google Scholar] [CrossRef]
- Wang, J.; Wu, Y.; Ma, C.; Fiorin, G.; Wang, J.; Pinto, L.H.; Lamb, R.A.; Klein, M.L.; DeGrado, W.F. Structure and inhibition of the drug-resistant S31N mutant of the M2 ion channel of influenza A virus. Proc. Natl. Acad. Sci. USA 2013, 110, 1315–1320. [Google Scholar] [CrossRef]
- Beniston, E.; Skittrall, J.P. Locations and structures of influenza A virus packaging-associated signals and other functional elements via an in silico pipeline for predicting constrained features in RNA viruses. PLoS Comput. Biol. 2024, 20, e1012009. [Google Scholar] [CrossRef]
- Elli, S.; Raffaini, G.; Guerrini, M.; Pond, S.K.; Matrosovich, M. Molecular modeling and phylogenetic analyses highlight the role of amino acid 347 of the N1 subtype neuraminidase in influenza virus host range and interspecies adaptation. Front. Microbiol. 2023, 14, 1309156. [Google Scholar] [CrossRef] [PubMed]
- Ford, C.T.; Yasa, S.; Obeid, K.; Jaimes, R.; Tomezsko, P.J.; Guirales-Medrano, S.; White, R.A.; Janies, D. Large-scale computational modelling of H5 influenza variants against HA1-neutralising antibodies. EBioMedicine 2025, 114, 105632. [Google Scholar] [CrossRef]
- Morgan, O.; Pebody, R. The WHO Hub for Pandemic and Epidemic Intelligence; supporting better preparedness for future health emergencies. Eurosurveillance 2022, 27, 2200385. [Google Scholar] [CrossRef]
- Kraemer, M.U.G.; Tsui, J.L.-H.; Chang, S.Y.; Lytras, S.; Khurana, M.P.; Vanderslott, S.; Bajaj, S.; Scheidwasser, N.; Curran-Sebastian, J.L.; Semenova, E.; et al. Artificial intelligence for modelling infectious disease epidemics. Nature 2025, 638, 623–635. [Google Scholar] [CrossRef]
- Domingo, E.; García-Crespo, C.; Lobo-Vega, R.; Perales, C. Mutation Rates, Mutation Frequencies, and Proofreading-Repair Activities in RNA Virus Genetics. Viruses 2021, 13, 1882. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Li, Y.; Yu, S.; Liu, L.; Luo, T.; Pu, Z.; Xiang, D.; Shen, X.; Irwin, D.M.; Liao, M.; et al. Adaptive evolution of human-isolated H5Nx avian influenza A viruses. Front. Microbiol. 2019, 10, 450518. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.M.L.; Komarasamy, T.V.; Adnan, N.A.A.B.; Radhakrishnan, A.K.; Balasubramaniam, V.R.M.T. Recent Advances, Approaches and Challenges in the Development of Universal Influenza Vaccines. Influenza Other Respir. Viruses 2024, 18, e13276. [Google Scholar] [CrossRef]
- Zhou, W.; Jiang, L.; Liao, S.; Wu, F.; Yang, G.; Hou, L.; Liu, L.; Pan, X.; Jia, W.; Zhang, Y. Vaccines’ New Era-RNA Vaccine. Viruses 2023, 15, 1760. [Google Scholar] [CrossRef]
- Attwaters, M. Broad protection from a single flu vaccine. Nat. Rev. Microbiol. 2023, 21, 67. [Google Scholar] [CrossRef]
- Knyazev, S.; Hughes, L.; Skums, P.; Zelikovsky, A. Epidemiological data analysis of viral quasispecies in the next-generation sequencing era. Brief. Bioinform. 2021, 22, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Vashisht, V.; Vashisht, A.; Mondal, A.K.; Farmaha, J.; Alptekin, A.; Singh, H.; Ahluwalia, P.; Srinivas, A.; Kolhe, R. Genomics for Emerging Pathogen Identification and Monitoring: Prospects and Obstacles. BioMedInformatics 2023, 3, 1145–1177. [Google Scholar] [CrossRef]
- Suminda, G.G.D.; Bhandari, S.; Won, Y.; Goutam, U.; Pulicherla, K.K.; Son, Y.-O.; Ghosh, M. High-throughput sequencing technologies in the detection of livestock pathogens, diagnosis, and zoonotic surveillance. Comput. Struct. Biotechnol. J. 2022, 20, 5378. [Google Scholar] [CrossRef]
- Bhatia, B.; Sonar, S.; Khan, S.; Bhattacharya, J. Pandemic-Proofing: Intercepting Zoonotic Spillover Events. Pathogens 2024, 13, 1067. [Google Scholar] [CrossRef]
- Ma, Y.; Frutos-Beltrán, E.; Kang, D.; Pannecouque, C.; De Clercq, E.; Menéndez-Arias, L.; Liu, X.; Zhan, P. Medicinal chemistry strategies for discovering antivirals effective against drug-resistant viruses. Chem. Soc. Rev. 2021, 50, 4514–4540. [Google Scholar] [CrossRef]
- Lamotte, L.A.; Tafforeau, L. How Influenza A Virus NS1 Deals with the Ubiquitin System to Evade Innate Immunity. Viruses 2021, 13, 2309. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Feng, Z.; Chen, Y.; Yang, L.; Liu, J.; Li, X.; Liu, S.; Zhou, L.; Wei, H.; Gao, R.; et al. Mammalian-Adaptive Mutation NP-Q357K in Eurasian H1N1 Swine Influenza Viruses Determines the Virulence Phenotype in Mice. Emerg. Microbes Infect. 2019, 8, 989–999. [Google Scholar] [CrossRef]
- Yamayoshi, S.; Yamada, S.; Fukuyama, S.; Murakami, S.; Zhao, D.; Uraki, R.; Watanabe, T.; Tomita, Y.; Macken, C.; Neumann, G.; et al. Virulence-Affecting Amino Acid Changes in the PA Protein of H7N9 Influenza A Viruses. J. Virol. 2014, 88, 3127. [Google Scholar] [CrossRef]
- Shirleen Soh, Y.; Moncla, L.H.; Eguia, R.; Bedford, T.; Bloom, J.D. Comprehensive Mapping of Adaptation of the Avian Influenza Polymerase Protein PB2 to Humans. eLife 2019, 8, e45079. [Google Scholar] [CrossRef]
- Yin, Y.; Zhang, X.; Qiao, Y.; Wang, X.; Su, Y.; Chen, S.; Qin, T.; Peng, D.; Liu, X. Glycosylation at 11Asn on Hemagglutinin of H5N1 Influenza Virus Contributes to Its Biological Characteristics. Vet. Res. 2017, 48, 81. [Google Scholar] [CrossRef]
- Xu, Y.; Peng, R.; Zhang, W.; Qi, J.; Song, H.; Liu, S.; Wang, H.; Wang, M.; Xiao, H.; Fu, L.; et al. Avian-to-Human Receptor-Binding Adaptation of Avian H7N9 Influenza Virus Hemagglutinin. Cell Rep. 2019, 29, 2217–2228.e5. [Google Scholar] [CrossRef]
- Wang, C.; Lee, H.H.Y.; Yang, Z.F.; Mok, C.K.P.; Zhang, Z. PB2-Q591K Mutation Determines the Pathogenicity of Avian H9N2 Influenza Viruses for Mammalian Species. PLoS ONE 2016, 11, e0162163. [Google Scholar] [CrossRef]
- Xiong, X.; Coombs, P.J.; Martin, S.R.; Liu, J.; Xiao, H.; McCauley, J.W.; Locher, K.; Walker, P.A.; Collins, P.J.; Kawaoka, Y.; et al. Receptor Binding by a Ferret-Transmissible H5 Avian Influenza Virus. Nature 2013, 497, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.; Cardenas Garcia, S.; Liu, J.; Santos, J.; Carnaccini, S.; Geiger, G.; Ferreri, L.; Rajao, D.; Perez, D.R. Alternative Strategy for a Quadrivalent Live Attenuated Influenza Virus Vaccine. J. Virol. 2018, 92, e01025-18. [Google Scholar] [CrossRef]
- Tang, W.; Li, X.; Tang, L.; Wang, T.; He, G. Characterization of the Low-Pathogenic H7N7 Avian Influenza Virus in Shanghai, China. Poult. Sci. 2021, 100, 565–574. [Google Scholar] [CrossRef]
- Thompson, A.J.; Paulson, J.C. Adaptation of Influenza Viruses to Human Airway Receptors. J. Biol. Chem. 2021, 296, e013309. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Li, C.; Li, X.F.; Deng, Y.Q.; Jiang, T.; Zhang, N.N.; Zu, S.; Zhang, R.R.; Li, L.; Chen, X.; et al. Type-IInterferon-Inducible SERTAD3 Inhibits Influenza A Virus Replication by Blocking the Assembly of Viral RNA Polymerase Complex. Cell Rep. 2020, 33, e108342. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Qi, J.; Xiao, H.; Bi, Y.; Zhang, W.; Xu, Y.; Wang, F.; Shi, Y.; Gao, G.F. Avian-to-Human Receptor-Binding Adaptation by Influenza A Virus Hemagglutinin H4. Cell Rep. 2017, 20, 1201–1214. [Google Scholar] [CrossRef]
- Zhao, P.; Sun, L.; Xiong, J.; Wang, C.; Chen, L.; Yang, P.; Yu, H.; Yan, Q.; Cheng, Y.; Jiang, L.; et al. Semiaquatic Mammals Might Be Intermediate Hosts to Spread Avian Influenza Viruses from Avian to Human. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef]
- Scheibner, D.; Salaheldin, A.H.; Bagato, O.; Zaeck, L.M.; Mostafa, A.; Blohm, U.; Müller, C.; Eweas, A.F.; Franzke, K.; Karger, A.; et al. Phenotypic Effects of Mutations Observed in the Neuraminidase of Human Origin H5N1 Influenza A Viruses. PLoS Pathog. 2023, 19, e1011135. [Google Scholar] [CrossRef] [PubMed]
- Kode, S.S.; Pawar, S.D.; Tare, D.S.; Keng, S.S.; Hurt, A.C.; Mullick, J. A Novel I117T Substitution in Neuraminidase of Highly Pathogenic Avian Influenza H5N1 Virus Conferring Reduced Susceptibility to Oseltamivir and Zanamivir. Vet. Microbiol. 2019, 235, 21–24. [Google Scholar] [CrossRef]
- Riegger, D.; Hai, R.; Dornfeld, D.; Mänz, B.; Leyva-Grado, V.; Sánchez-Aparicio, M.T.; Albrecht, R.A.; Palese, P.; Haller, O.; Schwemmle, M.; et al. The Nucleoprotein of Newly Emerged H7N9 Influenza A Virus Harbors a Unique Motif Conferring Resistance to Antiviral Human MxA. J. Virol. 2015, 89, 2241–2252. [Google Scholar] [CrossRef]
- Ran, W.; Schön, J.; Ciminski, K.; Kraft, J.; Kessler, S.; Euchner, S.; Hoffmann, D.; Pohlmann, A.; Beer, M.; Schwemmle, M.; et al. Generation of an Attenuated Chimeric Bat Influenza A Virus Live-Vaccine Prototype. Microbiol. Spectr. 2022, 10, e0142422. [Google Scholar] [CrossRef]
- Pu, Z.; Xiang, D.; Li, X.; Luo, T.; Shen, X.; Murphy, R.W.; Liao, M.; Shen, Y. Potential Pandemic of H7N9 Avian Influenza A Virus in Human. Front. Cell. Infect. Microbiol. 2018, 8, 404391. [Google Scholar] [CrossRef]
- Pham, P.T.V.; Turan, K.; Nagata, K.; Kawaguchi, A. Biochemical Characterization of Avian Influenza Viral Polymerase Containing PA or PB2 Subunit from Human Influenza A Virus. Microbes Infect. 2018, 20, 353–359. [Google Scholar] [CrossRef]
- Nguyen, N.M.; Sung, H.W.; Yun, K.J.; Park, H.; Yeo, S.J. Genetic Characterization of a Novel North American-Origin Avian Influenza A (H6N5) Virus Isolated from Bean Goose of South Korea in 2018. Viruses 2020, 12, 774. [Google Scholar] [CrossRef] [PubMed]
- Pawestri, H.A.; Nugraha, A.A.; Han, A.X.; Pratiwi, E.; Parker, E.; Richard, M.; van der Vliet, S.; Fouchier, R.A.M.; Muljono, D.H.; de Jong, M.D.; et al. Genetic and Antigenic Characterization of Influenza A/H5N1 Viruses Isolated from Patients in Indonesia, 2008–2015. Virus Genes 2020, 56, 417. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.L.; Wetzel, K.S.; Linderman, S.L.; Li, Y.; Sullivan, C.B.; Hensley, S.E. Compensatory Hemagglutinin Mutations Alter Antigenic Properties of Influenza Viruses. J. Virol. 2013, 87, 11168–11172. [Google Scholar] [CrossRef] [PubMed]
- Youk, S.-S.; Leyson, C.M.; Seibert, B.A.; Jadhao, S.; Perez, D.R.; Suarez, D.L.; Pantin-Jackwood, M.J. Mutations in PB1, NP, HA, and NA Contribute to Increased Virus Fitness of H5N2 Highly Pathogenic Avian Influenza Virus Clade 2.3.4.4 in Chickens. J. Virol. 2021, 95, e10-1128. [Google Scholar] [CrossRef]
- Lutz, M.; Schmierer, J.; Takimoto, T. Host Adaptive Mutations in the 2009 H1N1 Pandemic Influenza A Virus PA Gene Regulate Translation Efficiency of Viral MRNAs via GRSF1. Commun. Biol. 2022, 5, 1102. [Google Scholar] [CrossRef]
- Mair, C.M.; Ludwig, K.; Herrmann, A.; Sieben, C. Receptor Binding and PH Stability - How Influenza A Virus Hemagglutinin Affects Host-Specific Virus Infection. Biochim. Biophys. Acta Biomembr. 2014, 1838, 1153–1168. [Google Scholar] [CrossRef]
- Long, J.S.; Howard, W.A.; Núñez, A.; Moncorgé, O.; Lycett, S.; Banks, J.; Barclay, W.S. The Effect of the PB2 Mutation 627K on Highly Pathogenic H5N1 Avian Influenza Virus Is Dependent on the Virus Lineage. J. Virol. 2013, 87, 9983. [Google Scholar] [CrossRef]
- Liang, Y. Pathogenicity and Virulence of Influenza. Virulence 2023, 14. [Google Scholar] [CrossRef]
- Long, J.S.; Mistry, B.; Haslam, S.M.; Barclay, W.S. Host and Viral Determinants of Influenza A Virus Species Specificity. Nat. Rev. Microbiol. 2019, 17, 67–81. [Google Scholar] [CrossRef]
- Kobayashi, M.; Kodama, M.; Noshi, T.; Yoshida, R.; Kanazu, T.; Nomura, N.; Soda, K.; Isoda, N.; Okamatsu, M.; Sakoda, Y.; et al. Therapeutic Efficacy of Peramivir against H5N1 Highly Pathogenic Avian Influenza Viruses Harboring the Neuraminidase H275Y Mutation. Antiviral Res. 2017, 139, 41–48. [Google Scholar] [CrossRef]
- Kamiki, H.; Matsugo, H.; Kobayashi, T.; Ishida, H.; Takenaka-Uema, A.; Murakami, S.; Horimoto, T. A PB1-K577E Mutation in H9N2 Influenza Virus Increases Polymerase Activity and Pathogenicity in Mice. Viruses 2018, 10, 653. [Google Scholar] [CrossRef] [PubMed]
- Chrzastek, K.; Segovia, K.; Torchetti, M.; Killian, M.L.; Pantin-Jackwood, M.; Kapczynski, D.R. Virus Adaptation Following Experimental Infection of Chickens with a Domestic Duck Low Pathogenic Avian Influenza Isolate from the 2017 Usa H7n9 Outbreak Identifies Polymorphic Mutations in Multiple Gene Segments. Viruses 2021, 13, 1166. [Google Scholar] [CrossRef] [PubMed]
- Min, J.-Y.; Santos, C.; Fitch, A.; Twaddle, A.; Toyoda, Y.; DePasse, J.V.; Ghedin, E.; Subbarao, K. Mammalian Adaptation in the PB2 Gene of Avian H5N1 Influenza Virus. J. Virol. 2013, 87, 10884. [Google Scholar] [CrossRef] [PubMed]
- Hooper, K.A.; Bloom, J.D. A Mutant Influenza Virus That Uses an N1 Neuraminidase as the Receptor-Binding Protein. J. Virol. 2013, 87, 12531–12540. [Google Scholar] [CrossRef]
- Guo, Y.; Sun, T.; Bai, X.; Liang, B.; Deng, L.; Zheng, Y.; Yu, M.; Li, Y.; Ping, J. Comprehensive Analysis of the Key Amino Acid Substitutions in the Polymerase and NP of Avian Influenza Virus That Enhance Polymerase Activity and Affect Adaptation to Mammalian Hosts. Vet. Microbiol. 2023, 282, 109760. [Google Scholar] [CrossRef]
- Gao, R.; Cao, B.; Hu, Y.; Feng, Z.; Wang, D.; Hu, W.; Chen, J.; Jie, Z.; Qiu, H.; Xu, K.; et al. Human Infection with a Novel Avian-Origin Influenza A (H7N9) Virus. N. Engl. J. Med. 2013, 368, 1888–1897. [Google Scholar] [CrossRef]
- DesRochers, B.L.; Chen, R.E.; Gounder, A.P.; Pinto, A.K.; Bricker, T.; Linton, C.N.; Rogers, C.D.; Williams, G.D.; Webby, R.J.; Boon, A.C.M. Residues in the PB2 and PA Genes Contribute to the Pathogenicity of Avian H7N3 Influenza A Virus in DBA/2 Mice. Virology 2016, 494, 89–99. [Google Scholar] [CrossRef]
- Evseev, D.; Miranzo-Navarro, D.; Fleming-Canepa, X.; Webster, R.G.; Magor, K.E. Avian Influenza NS1 Proteins Inhibit Human, but Not Duck, RIG-I Ubiquitination and Interferon Signaling. J. Virol. 2022, 96, e00776-22. [Google Scholar] [CrossRef]
- de Vries, R.P.; Peng, W.; Grant, O.C.; Thompson, A.J.; Zhu, X.; Bouwman, K.M.; de la Pena, A.T.T.; van Breemen, M.J.; Ambepitiya Wickramasinghe, I.N.; de Haan, C.A.M.; et al. Three Mutations Switch H7N9 Influenza to Human-Type Receptor Specificity. PLoS Pathog. 2017, 13, e1006390. [Google Scholar] [CrossRef]
- Suttie, A.; Tok, S.; Yann, S.; Keo, P.; Horm, S.V.; Roe, M.; Kaye, M.; Sorn, S.; Holl, D.; Tum, S.; et al. Diversity of A(H5N1) Clade 2.3.2.1c Avian Influenza Viruses with Evidence of Reassortment in Cambodia, 2014–2016. PLoS ONE 2019, 14, e0226108. [Google Scholar] [CrossRef]
- Yu, Z.; Cheng, K.; Sun, W.; Zhang, X.; Li, Y.; Wang, T.; Wang, H.; Zhang, Q.; Xin, Y.; Xue, L.; et al. A PB1 T296R Substitution Enhance Polymerase Activity and Confer a Virulent Phenotype to a 2009 Pandemic H1N1 Influenza Virus in Mice. Virology 2015, 486, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Tzarum, N.; de Vries, R.P.; Peng, W.; Thompson, A.J.; Bouwman, K.M.; McBride, R.; Yu, W.; Zhu, X.; Verheije, M.H.; Paulson, J.C.; et al. The 150-Loop Restricts the Host Specificity of Human H10N8 Influenza Virus. Cell Rep. 2017, 19, 235. [Google Scholar] [CrossRef] [PubMed]
- Camacho-Zarco, A.R.; Kalayil, S.; Maurin, D.; Salvi, N.; Delaforge, E.; Milles, S.; Jensen, M.R.; Hart, D.J.; Cusack, S.; Blackledge, M. Molecular Basis of Host-Adaptation Interactions between Influenza Virus Polymerase PB2 Subunit and ANP32A. Nat. Commun. 2020, 11, 3656. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Mutation | Serotypes |
|---|---|---|
| PB1 | H99Y, K577E, N47S, Q694H, I695K, R486K, V709I, E391, G581, T661, G580, A660, T296R, E180D, M317V, R691K, 473R, L13P, 398Q, G70, V504, P69, S42, S375N, K52R, L212V | H5N1, H9N2, H7N4, H17N10, H18N11, H7N9, H2N2, H1N1, H3N2, H5N2, H5N8, H6N5 |
| PB2 | E627K, G590S, Q591R, H274Y, I222K, D701N, K683T, S155N, K526R, I382S, R389K, E120D, V227I, S265, F406Y, Q591K, L386V, V649I, I66M, I109V, I133V, E158G, G591R/K, V504, 615R, 558T, 714S, L13P, 398Q, G70, P69, S42, A274T, E129K, K702R, E543D, A655Y | H5N1, H9N2, H7N9, H7N4, H9N2, H17N10, H18N11, H7N7, H2N2, H1N1, H3N2, H5N2, H5N8, H6N5 |
| PB2 | E627K, G590S, Q591R, H274Y, I222K, D701N, K683T, S155N, K526R, I382S, R389K, E120D, V227I, S265, F406Y, Q591K, L386V, V649I, I66M, I109V, I133V, E158G, G591R/K, V504, 615R, 558T, 714S, L13P, 398Q, G70, P69, S42, A274T, E129K, K702R, E543D, A655Y | H5N1, H9N2, H7N9, H7N4, H9N2, H17N10, H18N11, H7N7, H2N2, H1N1, H3N2, H5N2, H5N8, H6N5 |
| PA | S388R, A448E, S49Y, D374G, T97I, I178M, M374T, V450A, E684G, F35L, K142E, P224S, L259P, I550L, T552S, M21I, S616P, E141K, N383D, M311I, R204K, P190S, Q400P, V127, L550, L627, 615R, 558T, L672, G18E, R388S, E448A, A36T, S37A, T85I, G186S, L336M, A343S/T, K356R, N409S | H5N1, H7N9, H6N1, H9N2, H1N1, H5N2, H17N10, H18N11, H7N7, H7N3, H5N8 |
| NP | N319K, I109T, Q357K, 105V, N52 | H5N1, H5N2, H1N1, H7N9 |
| HA | K165E, Q226L, N224K, G228S, V186G\K, K193T, A118T, S123N, A131V, R136K, L173I, M232I, H17, Y17, H106, H111, G225D, Q222L, G224S, G212R, N173H, N158D, T160A, T318I, H110Y, S138A, G186V, T221P, M66I, S141P, L322Q | H1N1, H2N2, H3N2, H5N1, H6N1, H10N8, H7N9, H10N4, H9N2 |
| NA | L204M, G147R, Deleted region residue 69 to 73, H274Y, I222K, H275Y, I117T, E368K, S416G, A46D, S319F, S430G | H3N2, H5N1, H7N9 |
| M2 | V27A, V27T, S31G, S31N | H5N1, H7N9 |
| NS1 | P42S, I176T, K217T | H5N1, H7N9, H5N2 |
| Mutation | Protein | Characteristic | Description |
|---|---|---|---|
| S388R | PA | Functional domain | Increases flexibility and associates with the vRNA promoter |
| A448E | PA | Functional domain | Forms hydrogen bonds with N444 interacting with PB1 and CDT POL II, enhancing the stability of PAC-C a16 helix |
| N409S | PA | Functional domain | Increases polymerase activity via interaction with PB-N |
| I382S | PB2 | Functional domain | Amino acid substitution in the PB2 cap-binding domain |
| L386V | PB2 | Functional domain | Mutation located within the cap-binding site |
| E190D, G225D | HA | Functional domain | Affects receptor-binding specificity (α2,3 SA, α2,6 SA) |
| G228S | HA | Functional domain | Forms a hydrogen bond with SA, increasing HA affinity for α2,6 SA |
| V186G-K193T-G228S/V186N-N224K-G228S | HA | Functional domain | Simultaneous substitutions for full α2,6 SA receptor specificity switch |
| N308S, A346V, T442A | NA | Functional impact | Residues near the active site of NA that may influence enzymatic activity |
| E368K, S416G | NA | Functional impact | Near the second sialic acid binding site, associated with neuraminidase activity and viral growth |
| K577E | PB1 | Functional impact | Conformational change in PB1 α-helix potentially affecting binding affinity for PB2 α-helix |
| Q694H, I695K | PB1 | Functional impact | Substitution in the PB1 C-terminal region |
| E627K | PB2 | Functional impact | Recruits a second polymerase for nascent vRNP formation |
| Q226L | HA | Functional impact | Establishes a hydrophobic environment complementary to α2,6 SA C6 atom, favoring human receptor binding |
| G212R, N173H | HA | Functional impact | May involve the globular head epitope-binding site of HA |
| N158D, T160A | HA | Functional impact | Causes the loss of a glycosylation site near the receptor-binding site, increasing the preference for human receptors |
| Host | Serotypes |
|---|---|
| Human | H9N9, H3N2, H1N1, H2N2, H7N9 |
| Chicken | H5N2, H7N9, H5N8, H9N2, H10N8, H4N6, H5N1, H7N3 |
| Dog | H3N2, H6N1 |
| Domestic cat | H5N6, H3N2 |
| Swine | H1N1, H2N2, H3N2, H5N1, H5N2, H9N2 |
| Turkey | H1N1, H2N2, H5N8, H5N2, H7N8, H7N3 |
| Swan | H5N1 |
| Goose | H4N8, H5N2, H5N6, H12N8, H7N7 |
| Seagull | H13N2, H13N8, H13N6, H13N9, H5N9 |
| Duck | H5N1, H5N2, H6N1, H6N2, H5N8, H3N8, H4N6, H7N1, H7N2, H7N3, H7N7, H13N6, H9N2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mellace, M.; Ceniti, C.; Cataldi, M.; Borrelli, L.; Tilocca, B. Avian Influenza Virus: Comparative Evolution as the Key for Predicting Host Tropism Expansion. Pathogens 2025, 14, 608. https://doi.org/10.3390/pathogens14070608
Mellace M, Ceniti C, Cataldi M, Borrelli L, Tilocca B. Avian Influenza Virus: Comparative Evolution as the Key for Predicting Host Tropism Expansion. Pathogens. 2025; 14(7):608. https://doi.org/10.3390/pathogens14070608
Chicago/Turabian StyleMellace, Matteo, Carlotta Ceniti, Marielda Cataldi, Luca Borrelli, and Bruno Tilocca. 2025. "Avian Influenza Virus: Comparative Evolution as the Key for Predicting Host Tropism Expansion" Pathogens 14, no. 7: 608. https://doi.org/10.3390/pathogens14070608
APA StyleMellace, M., Ceniti, C., Cataldi, M., Borrelli, L., & Tilocca, B. (2025). Avian Influenza Virus: Comparative Evolution as the Key for Predicting Host Tropism Expansion. Pathogens, 14(7), 608. https://doi.org/10.3390/pathogens14070608