1. Introduction
Induction of broadly cross-neutralizing antibodies remains a critical yet unresolved challenge in HIV-1 vaccine development. While 10–30% of infected individuals naturally develop bNAbs after years of chronic infection [
1,
2,
3,
4], this demonstrates that bNAb induction is biologically possible. Promisingly, Haynes et al. [
5] successfully induced polyclonal HIV-1 B cell lineages producing mature bNAbs by designing immunogens based on structural signatures of MPER-specific bNAbs, with the most potent antibodies neutralizing 15% of global tier-2 HIV-1 strains and 35% of clade B isolates. These breakthroughs highlight that focusing on structurally conserved functional epitopes—particularly those in key vulnerability regions—may be pivotal for effective bNAb induction.
Among these vulnerable targets, the V3-glycan supersites on HIV-1 Env represent key vulnerable regions targeted by bNAbs including PGT121, PGT128, and PGT135 [
6,
7]. Among these, the N332 glycan at the V3 loop base constitutes a major vulnerability site that enables broad antibody-mediated neutralization, making it a prime target for vaccine design [
8,
9]. While glycan loss at position 332 typically confers resistance to these bNAbs [
10], numerous circulating strains retain this supersite yet remain resistant to neutralization [
10,
11]. Emerging evidence suggests that V1/V2 loop elongation may mediate this resistance [
11,
12,
13,
14,
15], with van den Kerkhof et al. [
11] demonstrating a statistically significant correlation between V1 length and PGT135 resistance. Nevertheless, the precise structural and functional determinants of this resistance remain unclear, particularly given the known disparities between structurally defined and functionally relevant epitopes [
8,
16]. Elucidating these resistance mechanisms could provide critical insights for engineering improved HIV-1 Env immunogens capable of eliciting more effective neutralizing antibody responses.
Furthermore, the HIV-1 gp120 envelope glycoprotein exhibits a sophisticated structural organization comprising five variable (V1–V5) and five constant (C1–C5) regions that collectively enable both immune evasion and functional preservation [
17]. The surface-exposed variable regions undergo extensive sequence diversification to evade immune recognition [
18,
19,
20], while the internal constant regions maintain structural integrity through a conserved core scaffold [
21,
22]. This delicate equilibrium is maintained through extensive co-evolutionary networks across gp120 domains, where mutations in one region are often compensated by changes in another to retain overall functionality [
23,
24]. Understanding these intricate relationships provides critical insights for engineering antigenic modifications that maintain native-like conformation while eliciting bNAb responses.
In this study, we generated CBJC515-derived Env-pseudotyped viruses and characterized their neutralization profiles against a panel of bNAbs (PGT121, VRC01, 12A21, 10E8, 2G12, and PGT135), revealing a striking universal resistance to PGT135 despite preserved sensitivity to other bNAbs. Through site-directed mutagenesis and chimeric Env construction, we identified three novel resistance mechanisms: (i) V1 loop elongation, (ii) V4/C2 structural change, and (iii) acquisition of N398/N611 glycosylation sites, demonstrating HIV-1’s capacity for immune evasion without altering core epitopes. Importantly, our domain-swapping experiments uncovered critical interdomain compensatory mechanisms, showing that while V1V2 diversification broadly modulates Env functionality, the V3 region serves as a key stabilizer that rescues structural defects induced by V1V2 or C2 modifications [
25,
26], providing fundamental insights for designing next-generation immunogens targeting conserved neutralization epitopes.
2. Materials and Methods
2.1. Study Subjects
The study subjects CBJC515 and CBJC437 were selected from a well-characterized Chinese cohort of chronic HIV-1 subtype B’ infections, distinguished by their plasma’s broad cross-neutralizing activity against a diverse panel of 25 viral strains [
27]. These individuals acquired HIV-1 infection through commercial plasma donation between 1992–1995 and remained antiretroviral treatment (ART)-naïve throughout the study period. The major characteristics of CBJC515 are presented in
Table 1; those of CBJC437 have been previously documented [
28]. All study procedures were approved by the Institutional Review Board of the National Center for AIDS/STD Control and Prevention, China CDC, with written informed consent obtained from participants prior to blood collection and data acquisition.
2.2. Single-Genome Amplification and Env Clone Generation
Viral RNA was isolated from plasma samples using the QIAamp Viral RNA Mini Kit (Qiagen, Hessen, Germany), followed by immediate cDNA synthesis with the SuperScript III First-Strand Synthesis System (Invitrogen, Carlsbad, CA, USA). Full-length gp160 genes were amplified by single-genome amplification (SGA) as previously established [
29]. Briefly, diluted cDNA was distributed into replicate PCR reactions in 96-well plates (Thermo Fisher Scientific, Waltham, MA, USA), with endpoint dilution performed to achieve a positivity rate below 30%, ensuring single-template amplification in most wells.
The SGA-derived PCR products were cloned into the vector pcDNA3.1D/V5-His-TOPO following the manufacturer’s instructions (Invitrogen, Carlsbad, CA, USA). The correct pcDNA3.1-Env plasmids used to produce pseudoviruses were selected by sequencing. First, the constructed plasmids were transformed into E. coli JM109 competent cells (Takara Bio, Shiga Prefecture, Japan), and then selecting monoclonal E. coli colonies on ampicillin-resistant LB medium (ampicillin concentration: 50 μg/mL) for sequencing. The verified correct E. coli clones were cultivated and then the plasmids were extracted using E.Z.N.A.® Plasmid Mini Kit (Omega Bio-tek, Norcross, GA, USA) for pseudovirus production. The mutagenesis and chimeric plasmids described below, used to produce pseudoviruses, were selected as the same way.
2.3. Sequence Alignment and Bioinformatics Analysis
The SGA products were sequenced using an ABI 3700 Sequencer (Applied Biosciences, Foster City, CA, USA). Full-length
gp160 gene fragments were assembled and edited using Sequencher 4.1 (Gene Codes, Ann Arbor, MI, USA). All chromatograms were manually inspected for mixed bases (double peaks), and sequences exhibiting ambiguities were excluded from subsequent analyses. Env nucleotide sequences were aligned against the HIV-1 HXB2 reference strain using Gene Cutter (
https://www.hiv.lanl.gov/content/sequence/GENE_CUTTER/cutter.html, last accessed 16 February 2025), and corresponding amino acid (aa) sequences were derived from the nucleotide alignments. Potential N-linked glycosylation sites (PNGS) were predicted using N-Glycosite (
http://www.hiv.lanl.gov/content/sequence/GLYCOSITE/glycosite.html, last accessed 20 February 2025) from the Los Alamos HIV Database. Consensus sequences were generated using the online tool Consensus Maker (
https://www.hiv.lanl.gov/content/sequence/CONSENSUS/consensus.html, last accessed 21 October 2020). Sequence identity and similarity between the 2005 Env consensus and individual functional clones were computed using the LALIGN tool (
http://www.ch.embnet.org/software/LALIGN_form.html, last accessed 22 February 2025).
Additionally, a Chinese subtype B reference dataset (“B-Database”) was curated from the Los Alamos HIV Database (
https://www.hiv.lanl.gov/components/sequence/HIV/search/search.html, last accessed 10 March 2025) by applying the following filters:
China,
subtype B,
intact gp120 sequences, and
one sequence per patient. This dataset comprised 168 high-quality sequences for comparative analysis. Phylogenetic analysis was performed using MEGA software (version 5.1).
2.4. Construction of Site-Directed Mutants and Chimeric Env Clones
Site-directed mutagenesis was performed using a standard PCR-based approach in a 50 μL reaction system containing 50 ng plasmid DNA template, 1 μL each of 10 μM forward (F) and reverse (R) primers (Sangon Biotech, Shanghai, China), 4 μL dNTP mixture (2.5 mM each) (Takara Bio, Shiga Prefecture, Japan), 25 μL 2×PrimeSTAR GC buffer (Takara Bio, Shiga Prefecture, Japan), 0.5 μL PrimeSTAR HS DNA polymerase (2.5 U/μL; Takara Bio, Shiga Prefecture, Japan), and nuclease-free water to volume. The mutagenic primers were designed to include the nucleotide sequences corresponding to five amino acid residues flanking both sides of the target mutation site, with the reverse primer being fully complementary to the forward primer. The thermal cycling protocol consisted of an initial denaturation at 98 °C for 1 min, followed by 30 cycles of denaturation (98 °C for 10 s), annealing (56 °C for 20 s; temperature optimized based on primer Tm values), and extension (72 °C for 6 min), with a final extension at 72 °C for 10 min. The complete Env gene of each resulting mutant was subsequently sequenced to verify the intended mutations and ensure no unintended changes were introduced.
For construction of chimeric clones, the GeneArt
® Seamless Cloning and Assembly Kit (Invitrogen, Carlsbad, CA, USA) was utilized to assemble PCR-linearized vector backbones with custom-synthesized DNA fragments (Sangon Biotech, Shanghai, China). The Env plasmid backbone was amplified using primers containing 15 bp overlaps complementary to the insertion sequences, followed by verification via 0.8% agarose gel (Addgene, Watertown, MA, USA) electrophoresis and purification using the QIAquick Gel Extraction Kit (Qiagen, Hessen, Germany). The assembled chimeric plasmids were rigorously validated through both full-length sequencing and restriction enzyme digestion analysis. Functional validation of mutants and chimeric clones was performed by preparing pseudoviruses and assessing their infectivity as follows. The modified regions in the chimeric constructs were precisely defined according to HXB2 gp160 reference positions: V1 (aa 131–157), V2 (aa 158–196), V1V2 (aa 131–196), C2 (aa 197–295), V3 (aa 296–331), V4 (aa 385–418), and V1–V3 (aa 131–331). These domain boundaries are consistent with established HIV-1 envelope protein topology [
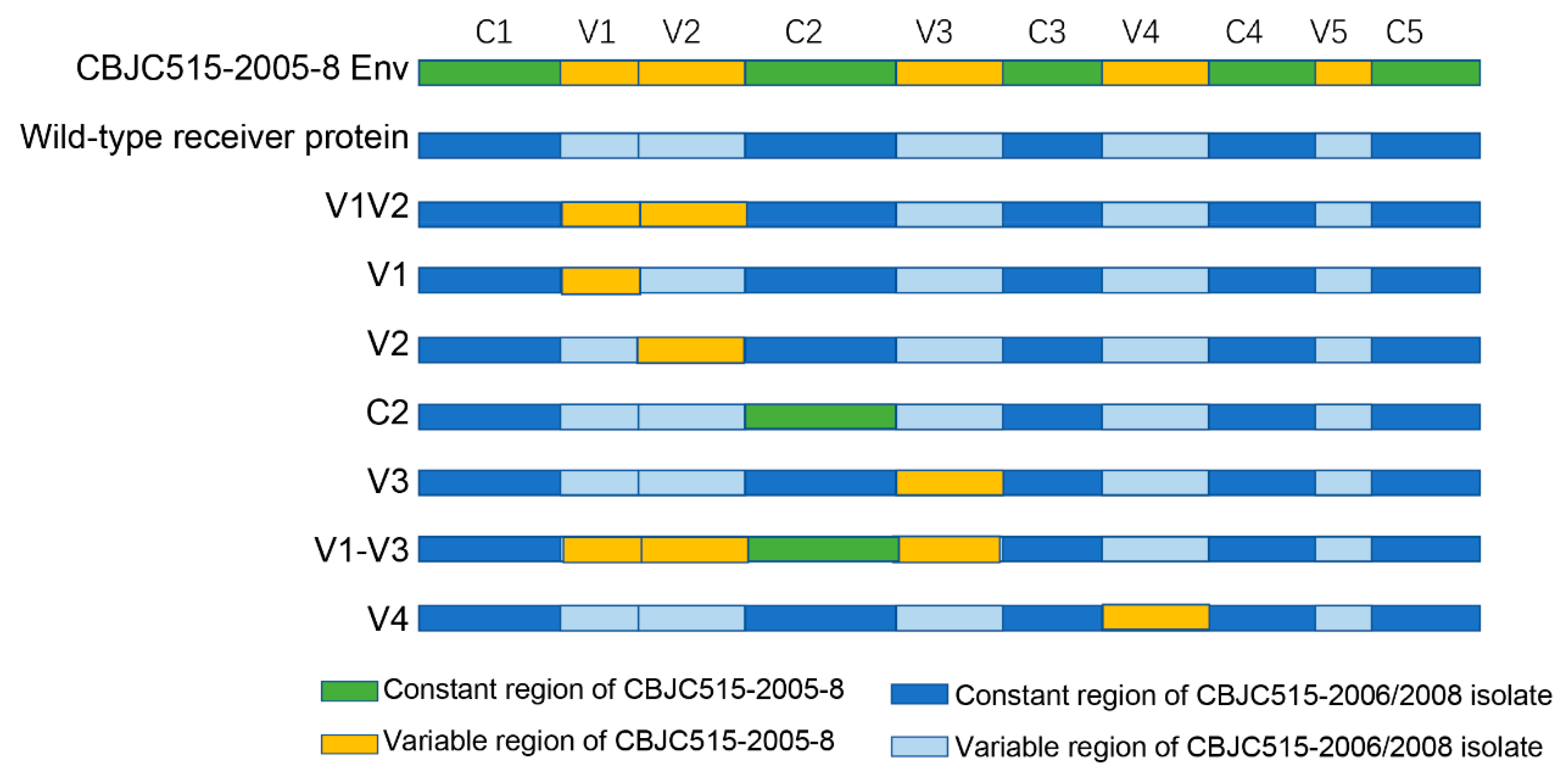
30], ensuring accurate domain swapping while maintaining structural integrity of the engineered Env proteins.
2.5. Pseudovirus Preparation and Titration
Pseudoviruses were generated by co-transfecting 4 × 106 293T/17 cells in a 75 cm² flask (Corning, New York, USA) with 5 μg Env expression plasmid (A260/A280 = 1.9) and 10 μg pSG3ΔEnv backbone vector using linear polyethylenimine (PEI Max, Polysciences, Warrington, PA, USA) at an N/P ratio of 7:1 (DNA:PEI ratio = 1:2 w/w). The PEI-DNA complexes were formed in serum-free DMEM (Life Technologies, Carlsbad, CA, USA) by incubating for 20 min at room temperature before addition to the cells. At 48 hours post-transfection, virus-containing supernatants were harvested, filtered through 0.45 μm membranes, and stored as 1 mL aliquots at −80 °C.
Pseudovirus infectivity was quantified using TZM-bl cells in a standardized TCID50 assay [
28]. Each well of 96-well culture plates was filled with 100 μL DMEM medium, followed by the addition of 25 μL pseudovirus stock to the first column with thorough mixing. A 5-fold serial dilution was then performed by transferring 25 μL from column to column through column 11 (with final 25 μL discarded from column 11), while column 12 was maintained as virus-free cell control. This entire dilution series was prepared in quadruplicate. Then 1 × 10
4 TZM-bl cells (containing DEAE-dextran, with a final concentration of 10 μg/mL) were added to each well and incubated for 48 hours in a cell culture incubator. Following incubation, 150 μL of supernatant was carefully removed from each well before adding 100 μL of Ultra-High Sensitivity Luminescence Reporter Gene Assay System (PerkinElmer, Waltham, MA, USA). After complete cell lysis, lysates were transferred to a black 96-well plate for luminescence measurement. Wells demonstrating relative luminescence units (RLUs) exceeding three-fold background (cell control) levels were considered positive for infection. The TCID
50 titers were determined using four independent replicates for each virus dilution series, with each replicate including technical duplicates. The final TCID
50 values represent the geometric mean (±SD) across these replicates, calculated using the Reed–Muench method implemented in the TCID
50 Macro for Microsoft Excel, provided by the Comprehensive Antibody-Vaccine Immune Monitoring Consortium (CAVIMC). This standardized computational tool ensures reproducible quantification of viral infectivity.
2.6. Neutralization Assay
The neutralization activity was assessed by measuring the reduction in luciferase reporter gene expression following a single-round infection of TZM-bl cells, as previously described [
27]. Pseudoviruses were normalized to 200 TCID
50, and 50 μL aliquots were incubated with 100 μL of threefold serially diluted bNAbs (in duplicate) for 1 h at 37 °C in 96-well flat-bottom plates (Corning, New York, NY, USA). The bNAb starting concentration was 20 μg/mL. Subsequently, the virus-antibody mixtures were used to infect 1 × 10
4 TZM-bl cells (100 μL/well) in the presence of DEAE-dextran (final concentration: 10 μg/mL). The 96-well plate was prepared with two control groups: column 1 served as a cell control with 150 μL DMEM and 100 μL TZM cells per well, while column 2 functioned as a virus control containing 100 μL DMEM, 50 μL viral suspension, and 100 μL TZM cells in each well. Following 48 h of incubation, infection levels were quantified by measuring luciferase activity. For luminescence measurement, 150 μL of culture supernatant was carefully removed from each well, and 100 μL of Ultra-High Sensitivity Luminescence Reporter Gene Assay System was added for 2 min to achieve complete cell lysis. Then, a 150 μL aliquot of each lysate was transferred to a black 96-well plate, and luminescence was measured using a PerkinElmer luminometer (PerkinElmer, Waltham, MA, USA). The 50% inhibitory dose was calculated as the antibody concentration that reduced relative luminescence units (RLU) by 50% compared to virus control wells (set as 100% infection).
2.7. Neutralizing Antibodies and Cell Lines
The bNAbs PGT135, PGT121, 2G12, 12A21, 10E8, and VRC01 were generously provided by the NIH AIDS Research and Reference Reagent Program. 293T/17 cells were purchased from the American Type Culture Collection (catalog no. 11268). The 293T/17 cell line is a derivative strain capable of generating high-titer infectious retroviruses. TZM-bl cells were obtained from the NIH AIDS Research and Reference Reagent Program (ARRRP, catalog no. 8129).
2.8. Statistical Analyses
GraphPad Prism 9.0 was used to perform statistical analyses and graphical representation. The IC50 of the antibody was calculated in GraphPad Prism by fitting a dose–response curve with a four-parameter logistic model, using nonlinear regression to determine the antibody concentration that inhibits 50% of viral activity.
4. Discussion
The induction of bNAb responses remains a central challenge in HIV vaccine development. Understanding escape mechanisms in broadly cross-neutralizing samples could provide critical insights for immunogen design [
15,
36,
37,
38]. Our study reveals multiple unconventional pathways of PGT135 escape, with the most significant novelty being the demonstration of epitope-independent resistance mechanisms in HIV-1. While canonical escape via N332 glycan loss was observed in early strains (2005), later strains (2006/2008) evolved resistance while preserving all critical PGT135 contact residues (N332/N386/N392/H330). This challenges the prevailing paradigm that bNAb escape necessarily requires epitope alteration and establishes three previously unrecognized evasion strategies: (1) V1 loop elongation, (2) V4/C2 structural change, and (3) acquisition of N398/N611 glycosylation sites. While previous work hypothesized that V1 elongation mediates escape [
11], we experimentally demonstrated that V1 replacement can restore sensitivity in some strains, whereas V1V2 or V2 substitutions cannot. These findings, combined with prior studies on PGT121 and PGT128 [
10,
13], underscore the broad impact of V1 length on V3-glycan bNAb neutralization, likely by sterically hindering access to the V3-glycan supersite [
15,
39]. We also identified additional roles for V4 and C2 regions in neutralization resistance. Through extensive mutagenesis, we found that single amino acid changes rarely restore sensitivity, suggesting that extreme variations may be required for PGT135 escape in N332
+ strains [
14]. The N398 residue is positioned within the V4 region, which has been implicated in modulating viral neutralization sensitivity [
6]. N611 is situated within the gp41 region, proximal to the fusion peptide of gp41. Studies indicate that N611 serves as a critical neutralizing epitope, and the N611A mutation has been shown to markedly enhance antibody neutralization efficacy [
40]. However, the mechanistic impact of the N398A and N611A mutations on the neutralization capacity of PGT135 remains unclear and warrants further investigation.
In donor CBJC515, we observed dynamic evolution of PNGS patterns, with N332 glycosylation sites emerging and becoming fixed by the 2006/2008 timepoints. While the N332 mutation in CBJC515-2005 strains represents the canonical escape route from V3-glycan bNAbs [
10], the persistence of this epitope in later strains suggests competing selection pressures from strain-specific NAbs or other antibody lineages [
15,
36]. This may have driven the virus to adopt alternative escape strategies while maintaining the N332 site. The fluctuating neutralization breadth from 2005–2009 (
Table 1) and evidence of coexisting antibody lineages [
41] support this hypothesis. In natural HIV-1 infection, the development of bNAbs typically requires several years of viral evolution and antigen exposure [
37,
42,
43]. This prolonged maturation process reflects both the need for extensive antibody somatic hypermutation and the complex structural features of conserved envelope epitopes that must be recognized. The retention of N332 through unconventional escape mechanisms may prolong epitope exposure, potentially facilitating bNAb maturation [
15,
36,
37,
38], as suggested by our observations of incomplete viral escape (data not shown). Further investigation through plasma epitope mapping and B-cell sequencing could elucidate these unusual escape patterns.
Our study reveals how HIV-1 maintains the delicate balance between immune evasion and viral fitness through coordinated evolution of envelope protein domains. While gp120’s hypervariable regions (particularly V1V2) permit extensive sequence diversification, our chimeric experiments demonstrate their functional dependence on co-evolved structural partners—evidenced by loss of Env functionality in 5/7 V1V2, 6/11 V1, and 2/3 V2 replacements (
Table 6,
Figure 3B). Similar constraints were observed in V4 (1/3 nonfunctional) and C2 (1/4 nonfunctional) chimeras (
Table 7,
Figure 3B), highlighting how HIV-1 preserves infectivity through precisely coordinated variations across structurally linked regions. These findings provide experimental validation of the “constrained diversification” model [
25,
26], wherein the virus accumulates immune-escape mutations while maintaining critical functional networks through compensatory coevolution.
Consistent with previous reports [
26], our chimeric studies demonstrated that V3 region replacement had minimal impact on Env functionality, with all V3-modified clones remaining functional (
Figure 3A). This remarkable tolerance to substitution likely stems from the V3 loop’s high conservation, which preserves the structural integrity of the co-receptor binding site [
44]. Notably, when we replaced the entire V1-V3 region (encompassing V1, V2, C2, and V3 domains), only 2 of 12 chimeras lost functionality (
Figure 3B), a significantly lower failure rate than observed with individual V1, V2, or C2 replacements. This finding suggests both coordinated evolution among these regions and a unique capacity of V3 to facilitate functional recovery. A striking example was observed in the CBJC515-2006-4 strain: while individual V1V2 or C2 replacements abrogated function, the complete V1-V3 chimera restored viability. These results align with recent studies demonstrating V3’s role in maintaining genetic robustness by compensating for structural perturbations [
25,
45].
Our findings significantly advance the understanding of PGT135 resistance mechanisms, revealing that even strains retaining key epitopes can evade neutralization through diverse modifications—including V1/V4/C2 alterations and N398/N611 glycosylation. These unconventional escape routes offer two key translational implications: (1) regarding vaccine design, it necessitates targeting conserved envelope regions beyond the primary epitope to block compensatory structural adaptations; (2) regarding antibody therapy, it demands combinatorial approaches that simultaneously address epitope variation and conformational shielding.
Additionally, molecular dynamics (MD) simulations have been widely employed to investigate protein-protein interactions and can be effectively applied to study the evolutionary mechanisms of viral membrane proteins. For instance, Han et al. utilized Gaussian accelerated molecular dynamics (GaMD) simulations combined with Markov state models to elucidate the inhibitory mechanism of fullerene-linear polyglycerol-b-aminosulfate (F-LGPS) on the spike protein [
46]. Similarly, Yu et al. demonstrated through homology modeling that VMP-63 and VMP-108 exhibit superior binding affinity with CBFβ [
47]. In future studies, we plan to enhance our investigations of interprotein molecular dynamics simulations to provide more comprehensive theoretical insights into viral evolutionary mechanisms.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}