
A Complete Transfer Learning-Based Pipeline for Discriminating Between Select Pathogenic Yeasts from Microscopy Photographs

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Culture Preparation
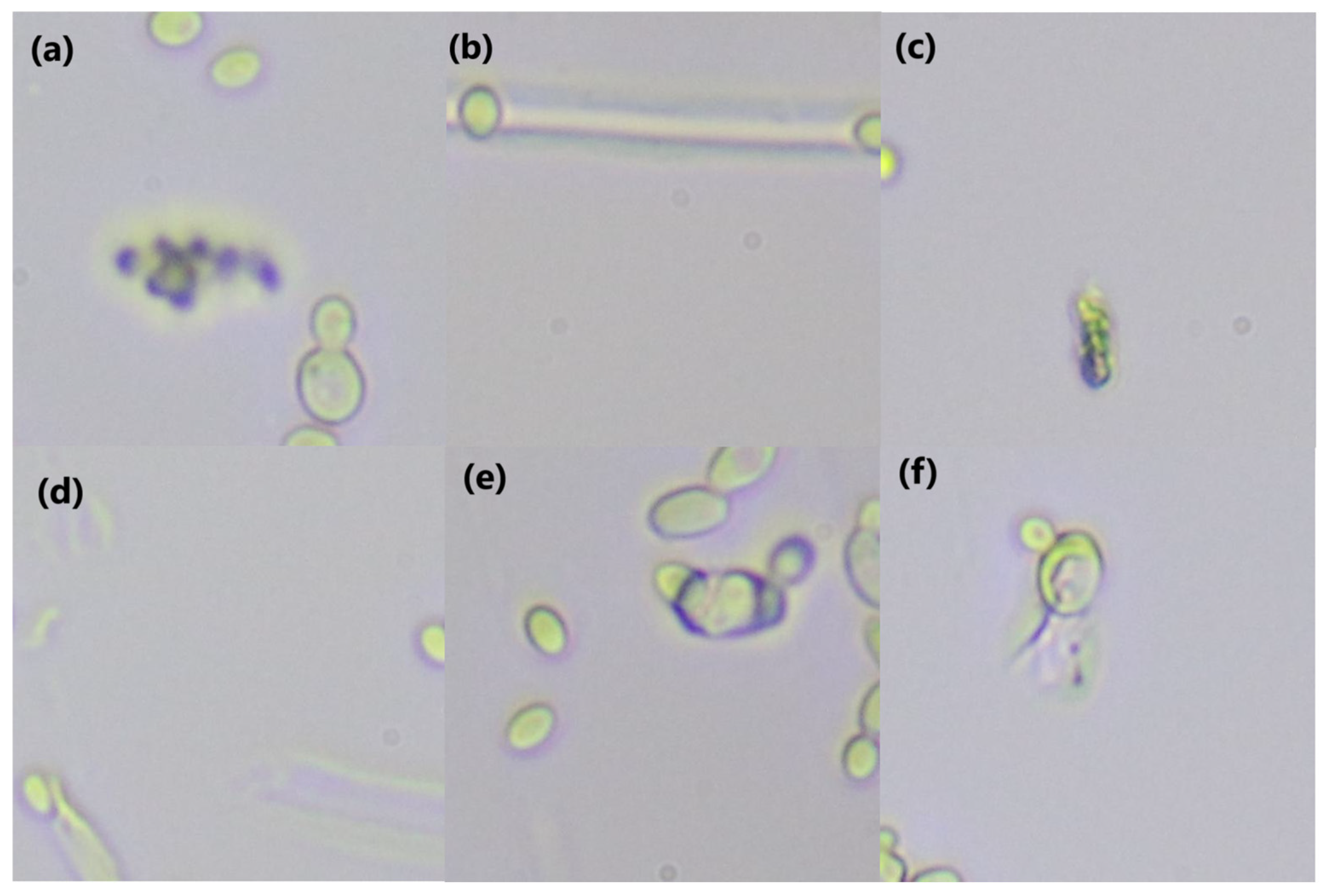
2.2. Microscope Slide Preparation and Imaging
2.3. Image Processing and Dataset
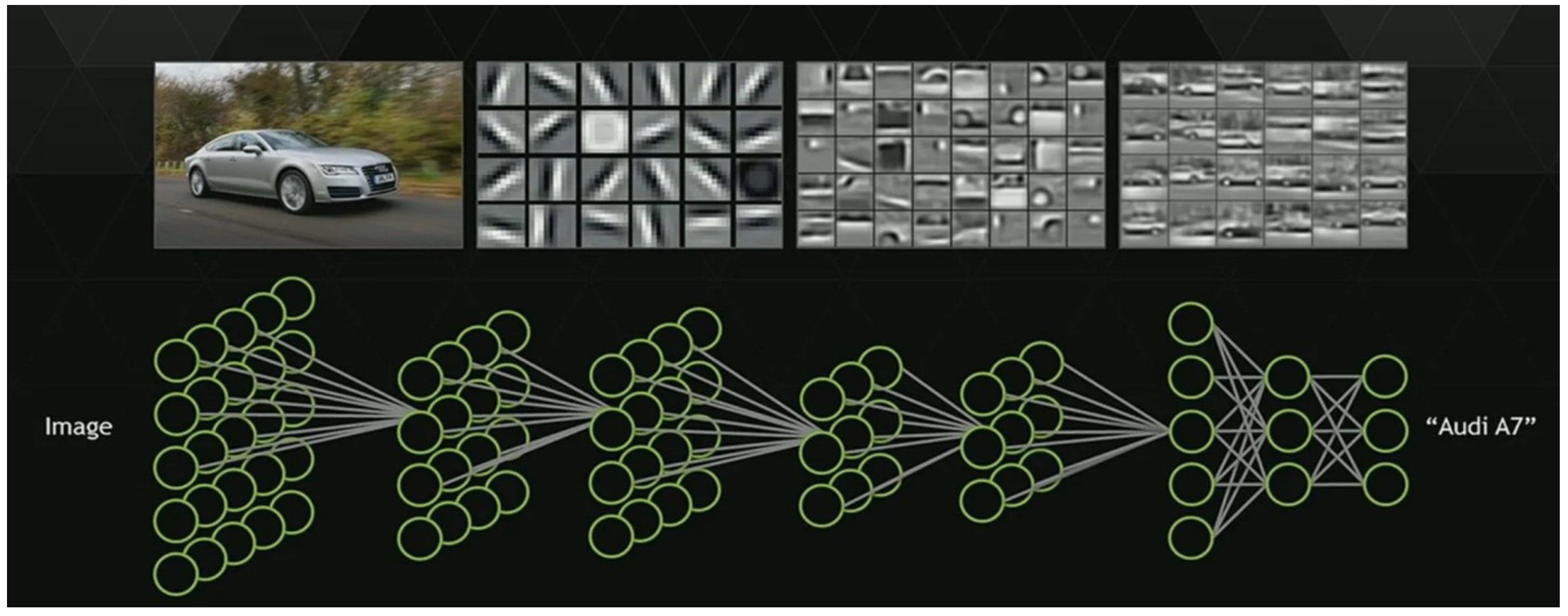
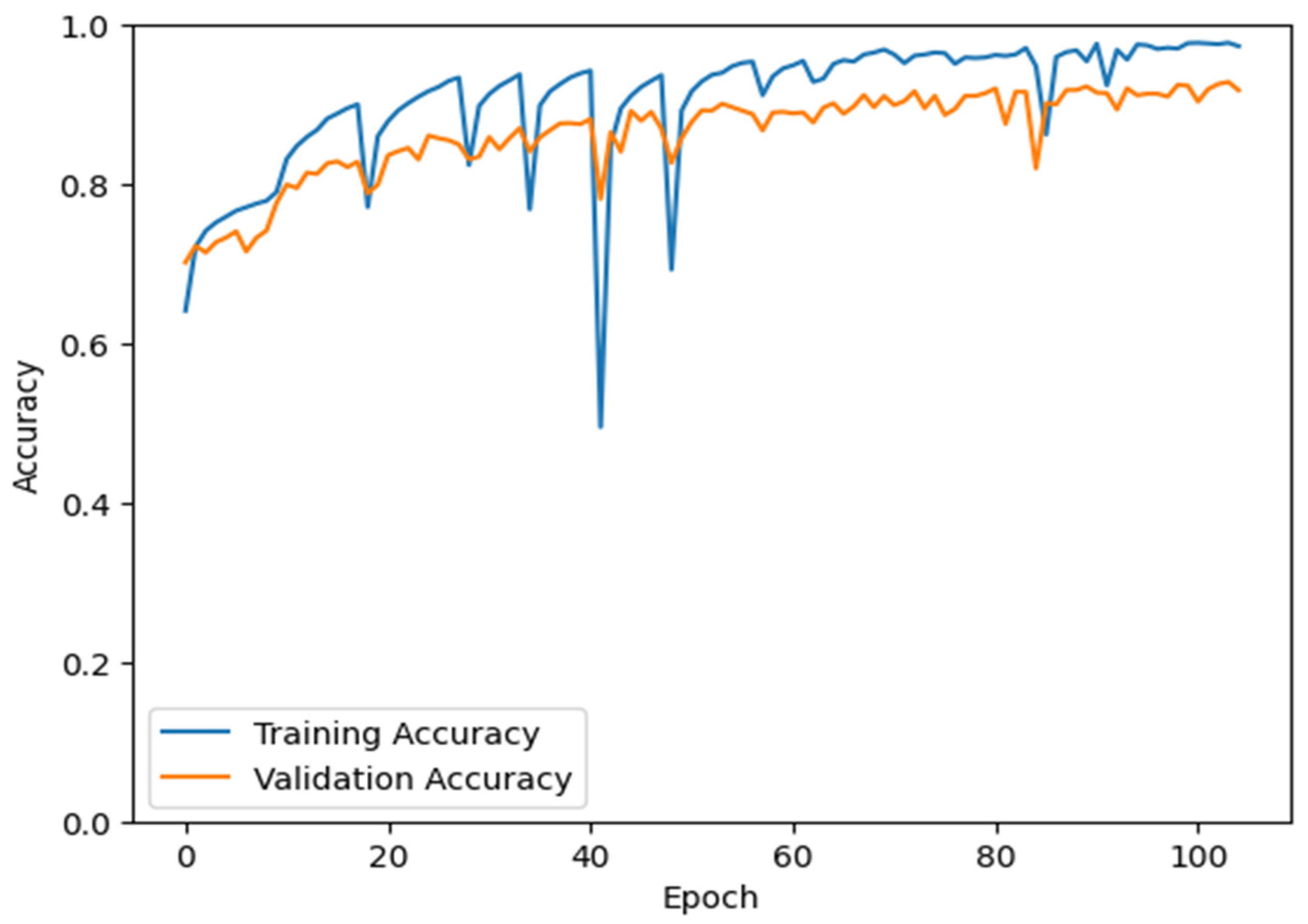
2.4. Model Training
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parker, R.A.; Gabriel, K.T.; Graham, K.; Butts, B.K.; Cornelison, C.T. Antifungal Activity of Select Essential Oils against Candida auris and Their Interactions with Antifungal Drugs. Pathogens 2022, 11, 821. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K.; Makimura, K.; Hasumi, Y.; Nishiyama, Y.; Uchida, K.; Yamaguchi, H. Candida auris sp. nov., a novel ascomycetous yeast isolated from the external ear canal of an inpatient in a Japanese hospital. Microbiol. Immunol. 2009, 53, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Kordalewska, M.; Perlin, D.S. Identification of Drug Resistant Candida auris. Front. Microbiol. 2019, 10, 1918. [Google Scholar] [CrossRef] [PubMed]
- Černáková, L.; Roudbary, M.; Brás, S.; Tafaj, S.; Rodrigues, C.F. Candida auris: A Quick Review on Identification, Current Treatments, and Challenges. Int. J. Mol. Sci. 2021, 22, 4470. [Google Scholar] [CrossRef] [PubMed]
- Jackson, B.R.; Chow, N.; Forsberg, K.; Litvintseva, A.P.; Lockhart, S.R.; Welsh, R.; Vallabhaneni, S.; Chiller, T. On the Origins of a Species: What Might Explain the Rise of Candida auris? J. Fungi 2019, 5, 58. [Google Scholar] [CrossRef] [PubMed]
- Fasciana, T.; Cortegiani, A.; Ippolito, M.; Giarratano, A.; Di Quattro, O.; Lipari, D.; Graceffa, D.; Giammanco, A. Candida auris: An Overview of How to Screen, Detect, Test and Control This Emerging Pathogen. Antibiotics 2020, 9, 778. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, Y.; Gao, Y.; Niu, X.; Li, J.; Tang, M.; Fu, C.; Qi, R.; Song, B.; Chen, H.; et al. Machine-learning based prediction of prognostic risk factors in patients with invasive candidiasis infection and bacterial bloodstream infection: A singled centered retrospective study. BMC Infect. Dis. 2022, 22, 150. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.T.; Xiao, J.; Sohn, M.B.; Fiscella, K.A.; Gilbert, C.; Grier, A.; Gill, A.L.; Gill, S.R. Machine learning approach identified Multi-Platform Factors for caries prediction in Child-Mother dyads. Front. Cell. Infect. Microbiol. 2021, 11, 727630. [Google Scholar] [CrossRef] [PubMed]
- CDC. Identification of Candida auris|Candida auris|Fungal Diseases. Available online: https://www.cdc.gov/fungal/candida-auris/identification.html (accessed on 28 August 2024).
- Fernández-Manteca, M.G.; Ocampo-Sosa, A.A.; Ruiz de Alegría-Puig, C.; Pía Roiz, M.; Rodríguez-Grande, J.; Madrazo, F.; Calvo, J.; Rodríguez-Cobo, L.; López-Higuera, J.M.; Fariñas, M.C.; et al. Automatic classification of Candida species using Raman spectroscopy and machine learning. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2023, 290, 122270. [Google Scholar] [CrossRef]
- Jeffery-Smith, A.; Taori, S.; Schelenz, S.; Jeffery, K.; Johnson, E.; Borman, A.M.; Candida auris Incident Management Team; Manuel, R.; Brown, C.S. Candida auris: A Review of the Literature. Clin. Microbiol. Rev. 2017, 31, e00029-17. [Google Scholar] [CrossRef] [PubMed]
- Russakovsky, O.; Deng, J.; Su, H.; Krause, J.; Satheesh, S.; Ma, S.; Huang, Z.; Karpathy, A.; Khosla, A.; Bernstein, A.; et al. ImageNet Large Scale Visual Recognition Challenge. Int. J. Comput. Vis. 2015, 115, 211–252. [Google Scholar] [CrossRef]
- Krizhevsky, A.; Hinton, G. Learning Multiple Layers of Features from Tiny Images. University of Toronto. 2009. Available online: https://www.cs.toronto.edu/~kriz/learning-features-2009-TR.pdf (accessed on 9 September 2023).
- Shaikh, J. Essentials of Deep Learning: Visualizing Convolutional Neural Networks in Python. Analytics Vidhya. 2018. Available online: https://www.analyticsvidhya.com/blog/2018/03/essentials-of-deep-learning-visualizing-convolutional-neural-networks (accessed on 1 April 2024).
- Lee, H.; Grosse, R.; Ranganath, R.; Ng, A.Y. Unsupervised learning of hierarchical representations with convolutional deep belief networks. Commun. ACM 2011, 54, 95–103. [Google Scholar] [CrossRef]
- CDC. How the AR Isolate Bank Helps Combat Antibiotic Resistance. Centers for Disease Control and Prevention. 2020. Available online: https://www.cdc.gov/drugresistance/resistance-bank/index.html (accessed on 8 August 2023).
- Welcome to the ARS Culture Collection (NRRL). ARS Culture Collection (NRRL). 2022. Available online: https://nrrl.ncaur.usda.gov/ (accessed on 8 August 2023).
- Li, L.; Jamieson, K.; DeSalvo, G.; Rostamizadeh, A.; Talwalkar, A. Hyperband: A novel Bandit-Based approach to hyperparameter optimization. arXiv 2016, arXiv:1603.06560. [Google Scholar]
- Simonyan, K.; Zisserman, A. Very deep convolutional networks for Large-Scale image recognition. arXiv 2014, arXiv:1409.1556. [Google Scholar]
- Howard, A.; Zhu, M.; Chen, B.; Kalenichenko, D.; Wang, W.; Weyand, T.; Andreetto, M.; Adam, A. MobileNets: Efficient convolutional neural networks for mobile vision applications. arXiv 2017, arXiv:1704.04861. [Google Scholar]
- Deng, J.; Dong, W.; Socher, R.; Li, L.; Li, K.; Li, F. ImageNet: A large-scale hierarchical image database. In Proceedings of the 2009 IEEE Conference on Computer Vision and Pattern Recognition, Miami, FL, USA, 20–25 June 2009. [Google Scholar] [CrossRef]
- Fukushima, K. Cognitron: A self-organizing multilayered neural network. Biol. Cybern. 1975, 20, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Džakula, N.B.; Bezdan, T. Convolutional Neural Network Layers and Architectures. In Proceedings of the Sinteza 2019—International Scientific Conference on Information Technology and Data Related Research, Novi Sad, Serbia, 20 April 2019. [Google Scholar] [CrossRef]
- Papers with Code—ReLU Explained. Available online: https://paperswithcode.com/method/relu (accessed on 28 August 2024).
- Howard, A.; Pang, R.; Adam, H.; Le, Q.V.; Sandler, M.; Chen, B.; Wang, W.; Chen, L.-C.; Tan, M.; Chu, G.; et al. Searching for MobileNetV3. In Proceedings of the 2019 IEEE/CVF International Conference on Computer Vision (ICCV), Seoul, Republic of Korea, 27 October–2 November 2019. [Google Scholar] [CrossRef]
- Papers with Code—Hard Swish Explained. Available online: https://paperswithcode.com/method/hard-swish (accessed on 28 August 2024).
- He, K.; Gkioxari, G.; Dollár, P.; Girshick, R. Mask R-CNN. In Proceedings of the 2017 IEEE International Conference on Computer Vision (ICCV), Venice, Italy, 22–29 October 2017. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identification Platform | Diagnosis of Candida auris |
|---|---|
| API 20C | Rhodotorula glutinis |
| Candida sake | |
| API ID 32C | Candida intermedia |
| Candida sake | |
| Saccharomyces kluyveri | |
| BD Phoenix Yeast Identification System | Candida haemulonii |
| Candida catenulata | |
| MicroScan | Candida famata |
| Candida lusitaniae | |
| Candida guilliermondii | |
| Candida parapsilosis | |
| RapID Yeast Plus | Candida parapsilosis |
| Vitek 2 YST | Candida haemulonii |
| Candida duobushaemulonii | |
| Vitek MS MALDI-TOF (with older libraries) | Candida lusitaniae |
| Candida haemulonii |
| Layer | Output Shape | Param # |
|---|---|---|
| InputLayer | (None, 224, 224, 3) | 0 |
| Conv2D | (None, 224, 224, 64) | 1792 |
| Conv2D | (None, 224, 224, 64) | 36,928 |
| MaxPooling2D | (None, 112, 112, 64) | 0 |
| Conv2D | (None, 112, 112, 128) | 73,856 |
| Conv2D | (None, 112, 112, 128) | 147,584 |
| MaxPooling2D | (None, 56, 56, 128) | 0 |
| Conv2D | (None, 56, 56, 256) | 295,168 |
| Conv2D | (None, 56, 56, 256) | 590,080 |
| Conv2D | (None, 56, 56, 256) | 590,080 |
| MaxPooling2D | (None, 28, 28, 256) | 0 |
| Conv2D | (None, 28, 28, 512) | 1,180,160 |
| Conv2D | (None, 28, 28, 512) | 2,359,808 |
| Conv2D | (None, 28, 28, 512) | 2,359,808 |
| MaxPooling2D | (None, 14, 14, 512) | 0 |
| Conv2D | (None, 14, 14, 512) | 0 |
| Conv2D | (None, 14, 14, 512) | 2,359,808 |
| Conv2D | (None, 14, 14, 512) | 2,359,808 |
| MaxPooling2D | (None, 7, 7, 512) | 0 |
| Layer | Output Shape | Param # |
|---|---|---|
| VGG16 Base Model | (None, 7, 7, 512) | 14,714,688 |
| Flatten | (None, 25088) | 0 |
| Dropout (50%) | (None, 25088) | 0 |
| Dense (256, PH-Swish) | (None, 256) | 6,422,784 |
| Dense (256, ReLU) | (None, 256) | 65,792 |
| Dropout (50%) | (None, 256) | 0 |
| Dense (128, PH-Swish) | (None, 128) | 32,896 |
| Output (6, Softmax) | (None, 6) | 774 |
| Model | Candida albicans | Candida auris | Candida glabrata | Candida haemulonii | Candida krusei | Saccharomyces cerevisiae | Overall |
|---|---|---|---|---|---|---|---|
| Hyperband CNN | 0.8866 | 0.8380 | 0.82364 | 0.8442 | 0.9249 | 0.8805 | 0.8652 |
| VGG16-Based CNN | 0.9544 | 0.9200 | 0.9529 | 0.9167 | 0.9428 | 0.9482 | 0.9391 |
| MobileNet-Based CNN | 0.7173 | 0.6931 | 0.6572 | 0.7386 | 0.8093 | 0.7865 | 0.7337 |
| Completed | 1.0000 | 1.0000 | 1.0000 | 0.9914 | 0.9636 | 0.9825 | 0.9909 |
| Pipeline (Whole Images) |
| Predicted Actual | Candida albicans | Candida auris | Candida glabrata | Candida haemulonii | Candida krusei | Saccharomyces cerevisiae |
|---|---|---|---|---|---|---|
| Candida albicans | 2447 | 7 | 48 | 17 | 46 | 93 |
| Candida auris | 1 | 2553 | 15 | 70 | 17 | 2 |
| Candida glabrata | 11 | 51 | 2466 | 115 | 3 | 12 |
| Candida haemulonii | 10 | 138 | 47 | 2431 | 24 | 8 |
| Candida krusei | 11 | 24 | 2 | 11 | 2589 | 21 |
| Saccharomyces cerevisiae | 84 | 2 | 10 | 8 | 67 | 2487 |
| Predicted Actual | Candida albicans | Candida auris | Candida glabrata | Candida haemulonii | Candida krusei | Saccharomyces cerevisiae |
|---|---|---|---|---|---|---|
| Candida albicans | 101 | 0 | 0 | 0 | 0 | 1 |
| Candida auris | 0 | 69 | 0 | 0 | 0 | 0 |
| Candida glabrata | 0 | 0 | 42 | 1 | 0 | 0 |
| Candida haemulonii | 0 | 0 | 0 | 115 | 0 | 0 |
| Candida krusei | 0 | 0 | 0 | 0 | 53 | 0 |
| Saccharomyces cerevisiae | 0 | 0 | 0 | 0 | 2 | 56 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parker, R.A.; Hannagan, D.S.; Strydom, J.H.; Boon, C.J.; Fussell, J.; Mitchell, C.A.; Moerschel, K.L.; Valter-Franco, A.G.; Cornelison, C.T. A Complete Transfer Learning-Based Pipeline for Discriminating Between Select Pathogenic Yeasts from Microscopy Photographs. Pathogens 2025, 14, 504. https://doi.org/10.3390/pathogens14050504
Parker RA, Hannagan DS, Strydom JH, Boon CJ, Fussell J, Mitchell CA, Moerschel KL, Valter-Franco AG, Cornelison CT. A Complete Transfer Learning-Based Pipeline for Discriminating Between Select Pathogenic Yeasts from Microscopy Photographs. Pathogens. 2025; 14(5):504. https://doi.org/10.3390/pathogens14050504
Chicago/Turabian StyleParker, Ryan A., Danielle S. Hannagan, Jan H. Strydom, Christopher J. Boon, Jessica Fussell, Chelbie A. Mitchell, Katie L. Moerschel, Aura G. Valter-Franco, and Christopher T. Cornelison. 2025. "A Complete Transfer Learning-Based Pipeline for Discriminating Between Select Pathogenic Yeasts from Microscopy Photographs" Pathogens 14, no. 5: 504. https://doi.org/10.3390/pathogens14050504
APA StyleParker, R. A., Hannagan, D. S., Strydom, J. H., Boon, C. J., Fussell, J., Mitchell, C. A., Moerschel, K. L., Valter-Franco, A. G., & Cornelison, C. T. (2025). A Complete Transfer Learning-Based Pipeline for Discriminating Between Select Pathogenic Yeasts from Microscopy Photographs. Pathogens, 14(5), 504. https://doi.org/10.3390/pathogens14050504