
First Animal Source Metagenome Assembly of Lawsonella clevelandensis from Canine External Otitis



Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction, Library Preparation, and Sequencing
2.3. Bioinformatic Analysis
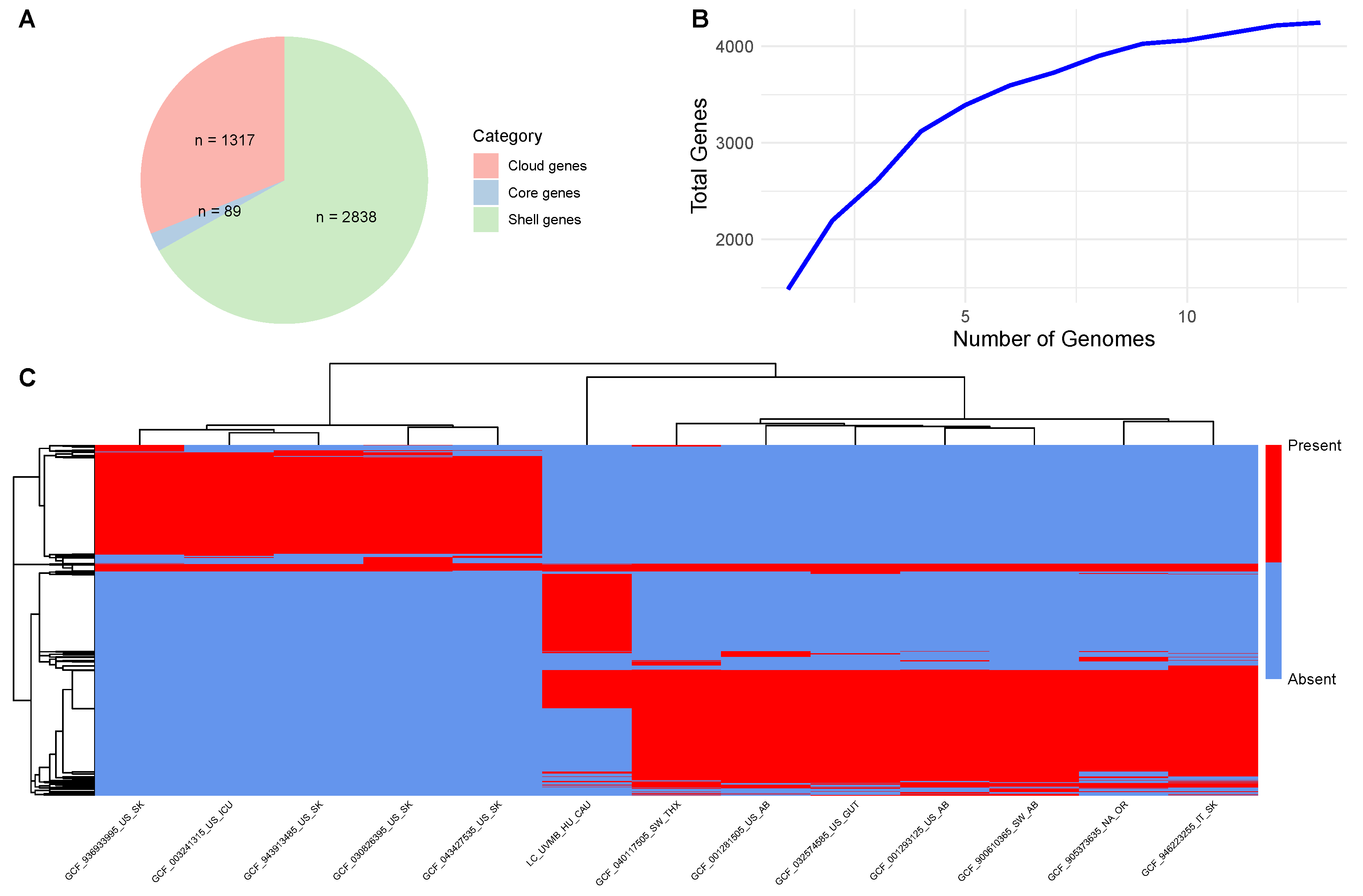
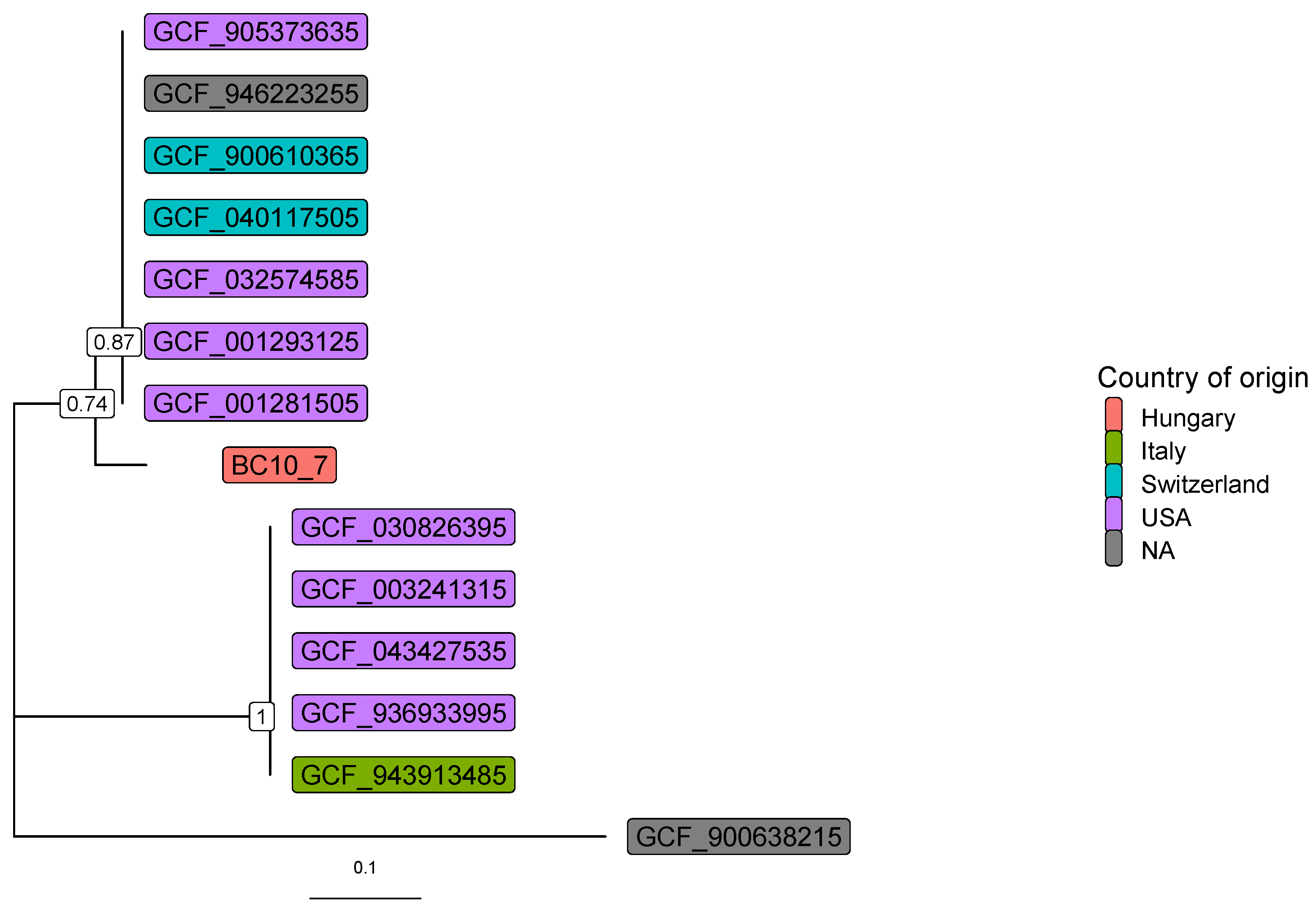
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Proverbio, D.; Perego, R.; Spada, E.; Ferro, E. Prevalence of adverse food reactions in 130 dogs in Italy with dermatological signs: A retrospective study. J. Small Anim. Pract. 2010, 51, 370–374. [Google Scholar] [CrossRef] [PubMed]
- White, R.; Pomeroy, C. Total ear canal ablation and lateral bulla osteotomy in the dog. J. Small Anim. Pract. 1990, 31, 547–553. [Google Scholar] [CrossRef]
- Chiu, C.Y.; Miller, S.A. Clinical metagenomics. Nat. Rev. Genet. 2019, 20, 341–355. [Google Scholar] [CrossRef]
- Sheka, D.; Alabi, N.; Gordon, P.M. Oxford nanopore sequencing in clinical microbiology and infection diagnostics. Briefings Bioinform. 2021, 22, bbaa403. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.E.; Bernard, K.A.; Harrington, S.M.; Patel, N.B.; Tucker, T.A.; Metcalfe, M.G.; McQuiston, J.R. Lawsonella clevelandensis gen. nov., sp. nov., a new member of the suborder Corynebacterineae isolated from human abscesses. Int. J. Syst. Evol. Microbiol. 2016, 66, 2929–2935. [Google Scholar] [CrossRef]
- Ramesh, R.; Assi, M.; Garrigos, Z.E.; Sohail, M.R. Lawsonella clevelandensis: An emerging cause of vascular graft infection. BMJ Case Rep. CP 2021, 14, e237350. [Google Scholar] [CrossRef] [PubMed]
- Nour, S.I.; Khodadadi, R.B.; Schuetz, A.N.; Patel, R.; Saleh, O.M.A. Lawsonella clevelandensis, a case series of vascular graft infections caused by a rare pathogen. IDCases 2023, 31, e01735. [Google Scholar] [CrossRef]
- De Coster, W.; D’hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI reference sequence (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007, 33, D61–D65. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Bickhart, D.M.; Behsaz, B.; Gurevich, A.; Rayko, M.; Shin, S.B.; Kuhn, K.; Yuan, J.; Polevikov, E.; Smith, T.P.; et al. metaFlye: Scalable long-read metagenome assembly using repeat graphs. Nat. Methods 2020, 17, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.W.; Simmons, B.A.; Singer, S.W. MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 2016, 32, 605–607. [Google Scholar] [CrossRef]
- Alneberg, J.; Bjarnason, B.S.; de Bruijn, I.; Schirmer, M.; Quick, J.; Ijaz, U.Z.; Loman, N.J.; Andersson, A.F.; Quince, C. CONCOCT: Clustering contigs on coverage and composition. arXiv 2013, arXiv:1312.4038. [Google Scholar]
- Wang, Z.; Huang, P.; You, R.; Sun, F.; Zhu, S. MetaBinner: A high-performance and stand-alone ensemble binning method to recover individual genomes from complex microbial communities. Genome Biol. 2023, 24, 1. [Google Scholar] [CrossRef]
- Liu, C.C.; Dong, S.S.; Chen, J.B.; Wang, C.; Ning, P.; Guo, Y.; Yang, T.L. MetaDecoder: A novel method for clustering metagenomic contigs. Microbiome 2022, 10, 46. [Google Scholar] [CrossRef]
- Wickramarachchi, A.; Lin, Y. LRBinner: Binning long reads in metagenomics datasets. In Proceedings of the 21st International Workshop on Algorithms in Bioinformatics (WABI 2021), Virtual, 2–4 September 2021; Schloss Dagstuhl–Leibniz-Zentrum für Informatik: Wadern, Germany, 2021; pp. 11:1–11:18. [Google Scholar]
- Pan, S.; Zhao, X.M.; Coelho, L.P. SemiBin2: Self-supervised contrastive learning leads to better MAGs for short-and long-read sequencing. Bioinformatics 2023, 39 (Suppl. 1), i21–i29. [Google Scholar] [CrossRef]
- Sieber, C.M.K.; Probst, A.J.; Sharrar, A.; Thomas, B.C.; Hess, M.; Tringe, S.G.; Banfield, J.F. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 2018, 3, 836–843. [Google Scholar] [CrossRef]
- Chaumeil, P.A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk v2: Memory friendly classification with the genome taxonomy database. Bioinformatics 2022, 38, 5315–5316. [Google Scholar] [CrossRef] [PubMed]
- Chklovski, A.; Parks, D.H.; Woodcroft, B.J.; Tyson, G.W. CheckM2: A rapid, scalable and accurate tool for assessing microbial genome quality using machine learning. Nat. Methods 2023, 20, 1203–1212. [Google Scholar] [CrossRef]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and taxonomy in diagnostics for food security: Soft-rotting enterobacterial plant pathogens. Anal. Methods 2016, 8, 12–24. [Google Scholar] [CrossRef]
- Chen, L.; Zheng, D.; Liu, B.; Yang, J.; Jin, Q. VFDB 2016: Hierarchical and refined dataset for big data analysis—10 years on. Nucleic Acids Res. 2016, 44, D694–D697. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- McArthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L.; et al. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 2013, 57, 3348–3357. [Google Scholar] [CrossRef]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Krawczyk, P.S.; Lipinski, L.; Dziembowski, A. PlasFlow: Predicting plasmid sequences in metagenomic data using genome signatures. Nucleic Acids Res. 2018, 46, e35. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef]
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 2021, 39, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef] [PubMed]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Samland, A.K.; Sprenger, G.A. Transaldolase: From biochemistry to human disease. Int. J. Biochem. Cell Biol. 2009, 41, 1482–1494. [Google Scholar] [CrossRef]
- Xu, S.; Li, L.; Luo, X.; Chen, M.; Tang, W.; Zhan, L.; Dai, Z.; Lam, T.T.; Guan, Y.; Yu, G. Ggtree: A serialized data object for visualization of a phylogenetic tree and annotation data. IMeta 2022, 1, e56. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Schliep, K.; Potts, A.J.; Morrison, D.A.; Grimm, G.W. Intertwining phylogenetic trees and networks. Methods Ecol. Evol. 2017, 8, 1212–1220. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2023; Available online: https://www.R-project.org/ (accessed on 25 April 2025).
- Woo, Y.R.; Cho, M.; Han, Y.; Lee, S.H.; Cho, S.H.; Lee, J.D.; Kim, H.S. Characterization of distinct microbiota associated with scalp dermatitis in patients with atopic dermatitis. J. Clin. Med. 2022, 11, 1735. [Google Scholar] [CrossRef] [PubMed]
- Lijia, Z.; Jia Chao, Q.; Li, L.; Shikun, D.; Peiyang, G. Case report: A case of widespread soft tissue infection and multiple abscesses secondary to hidradenitis suppurativa inducing septic shock caused by Lawsonella clevelandensis in China. Front. Med. 2024, 11, 1392430. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Zhang, J.; Gong, L.; Wang, G.; Khan, A.; Cui, H. Infection caused by Lawsonella clevelandensis after breast augmentation with autologous fat grafting: A case report. BMC Infect. Dis. 2023, 23, 124. [Google Scholar] [CrossRef]
- Favila Menezes, M.; Sousa, M.J.; Paixão, P.; Atouguia, J.; Negreiros, I.; Simões, M. Lawsonella clevelandensis as the causative agent of a breast abscess. IDCases 2018, 12, 95–96. [Google Scholar] [CrossRef]
- Chudy-Onwugaje, K.; Vandermeer, F.; Quezada, S. Mimicking abdominal tuberculosis: Abdominal abscess caused by Lawsonella clevelandensis in inflammatory bowel disease. Clin. Gastroenterol. Hepatol. 2019, 17, e92. [Google Scholar] [CrossRef]
- Kumaria, A.; Lucas, E.; Crusz, S.; Howarth, S.; Cartmill, M. Lawsonella clevelandensis causing spinal subdural empyema. Br. J. Neurosurg. 2023, 37, 653–655. [Google Scholar] [CrossRef] [PubMed]
- Morton, A.B.; Boyle, E.; Pettengill, M.A.; Gancher, E. The Brief Case: Strictly Anaerobic and Staining Acid Fast. J. Clin. Microbiol. 2023, 61, e00150-22. [Google Scholar] [CrossRef]
- Wallen, Z.D.; Appah, M.; Dean, M.N.; Sesler, C.L.; Factor, S.A.; Molho, E.; Zabetian, C.P.; Standaert, D.G.; Payami, H. Characterizing dysbiosis of gut microbiome in PD: Evidence for overabundance of opportunistic pathogens. npj Park. Dis. 2020, 6, 11. [Google Scholar] [CrossRef]
- Ahmed, W.; Dewar, S.; Williams, R.; Stansby, G.; Harris, K.; Weiand, D. Lawsonella clevelandensis is a rare cause of infected chronic contained rupture of abdominal aortic aneurysm. Access Microbiol. 2021, 3, 000183. [Google Scholar] [CrossRef]
- Kim, J.H.; Son, S.M.; Park, H.; Kim, B.K.; Choi, I.S.; Kim, H.; Huh, C.S. Taxonomic profiling of skin microbiome and correlation with clinical skin parameters in healthy Koreans. Sci. Rep. 2021, 11, 16269. [Google Scholar] [CrossRef]
- Escapa, I.F.; Chen, T.; Huang, Y.; Gajare, P.; Dewhirst, F.E.; Lemon, K.P. New insights into human nostril microbiome from the expanded human oral microbiome database (eHOMD): A resource for the microbiome of the human aerodigestive tract. Msystems 2018, 3, e00187-18. [Google Scholar] [CrossRef] [PubMed]
- Polak-Witka, K.; Constantinou, A.; Schwarzer, R.; Helmuth, J.; Wiessner, A.; Hadam, S.; Kanti, V.; Rancan, F.; Andruck, A.; Richter, C.; et al. Identification of anti-microbial peptides and traces of microbial DNA in infrainfundibular compartments of human scalp terminal hair follicles. Eur. J. Dermatol. 2021, 31, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Leung, M.; Tong, X.; Bøifot, K.O.; Bezdan, D.; Butler, D.J.; Danko, D.C.; Gohli, J.; Green, D.; Hernandez, M.T.; Kelly, F.J.; et al. Characterization of the public transit air microbiome and resistome reveals geographical specificity. Microbiome 2021, 9, 112. [Google Scholar] [CrossRef]
- Fritz, B.; März, M.; Weis, S.; Wahl, S.; Ziemssen, F.; Egert, M. Site-specific molecular analysis of the bacteriota on worn spectacles. Sci. Rep. 2020, 10, 5577. [Google Scholar] [CrossRef]
- Francuzik, W.; Franke, K.; Schumann, R.R.; Heine, G.; Worm, M. Propionibacterium acnes abundance correlates inversely with Staphylococcus aureus: Data from atopic dermatitis skin microbiome. Acta Dermato-Venereol. 2018, 98, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Song, J.W.; Lee, C.W.; Kim, D.S.; Sohn, J.; Lee, S. Skin Barrier-Enhancing Effects of Dermabiotics HDB with Regulation of Skin Microflora. J. Microbiol. Biotechnol. 2023, 34, 1–9. [Google Scholar]
- Cojkic, A.; Niazi, A.; Guo, Y.; Hallap, T.; Padrik, P.; Morrell, J.M. Identification of bull semen microbiome by 16S sequencing and possible relationships with fertility. Microorganisms 2021, 9, 2431. [Google Scholar] [CrossRef]
- Ozga, A.T.; Ottoni, C. Dental calculus as a proxy for animal microbiomes. Quat. Int. 2023, 653, 47–52. [Google Scholar] [CrossRef]
- Paul, L.J.; Ericsson, A.C.; Andrews, F.M.; McAdams, Z.; Keowen, M.L.; St Blanc, M.P.; Banse, H.E. Field study examining the mucosal microbiome in equine glandular gastric disease. PLoS ONE 2023, 18, e0295697. [Google Scholar] [CrossRef]
- Nicholson, A.C.; Bell, M.; Humrighouse, B.W.; McQuiston, J.R. Complete genome sequences for two strains of a novel fastidious, partially acid-fast, Gram-positive Corynebacterineae bacterium, derived from human clinical samples. Genome Announc. 2015, 3, 10–1128. [Google Scholar] [CrossRef]
- Goldenberger, D.; Naegele, M.; Steffens, D.; Eichenberger, R.; Egli, A.; Seth-Smith, H. Emerging anaerobic and partially acid-fast Lawsonella clevelandensis: Extended characterization by antimicrobial susceptibility testing and whole genome sequencing. Clin. Microbiol. Infect. 2019, 25, 1447–1448. [Google Scholar] [CrossRef]
- Leonard, C.; Picavet, P.P.; Fontaine, J.; Clercx, C.; Taminiau, B.; Daube, G.; Claeys, S. The Middle Ear Microbiota in Healthy Dogs Is Similar to That of the External Ear Canal. Vet. Sci. 2023, 10, 216. [Google Scholar] [CrossRef] [PubMed]
- Ring, N.; Low, A.S.; Wee, B.; Paterson, G.K.; Nuttall, T.; Gally, D.; Mellanby, R.; Fitzgerald, J.R. Rapid metagenomic sequencing for diagnosis and antimicrobial sensitivity prediction of canine bacterial infections. Microb. Genom. 2023, 9, 001066. [Google Scholar] [CrossRef] [PubMed]
- Petri, F.; Mahmoud, O.K.; Ranganath, N.; El Zein, S.; Abu Saleh, O.; Berbari, E.F.; Fida, M. Plasma Microbial Cell-free DNA Next-generation Sequencing Can Be a Useful Diagnostic Tool in Patients With Osteoarticular Infections. Open Forum Infect. Dis. 2024, 11, ofae328. [Google Scholar] [CrossRef]
- Zavadzki, G.; Reyes Barros, T.; Sarfatis Feige, A.; Granada Castaño, J.; Kobus Garin, V.; Ramos-Rojas, J.; Bigossi Aguiar, N.; Inzunza Robles, J. Reporte del primer caso de infección por Lawsonella clevelandensis en Latinoamérica y revisión de literatura. Rev. Médica Chile 2023, 151, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Yamada, A.; Masuda, H.; Kashiwazaki, E.; Nakayama, S.; Kadokura, T.; Sakai, K.; Tashiro, Y. Sample collecting methods for bacterial community structure analysis of scalp hair: Non-invasive swabbing versus intrusive hair shaft cutting. Sci. Rep. 2024, 14, 22461. [Google Scholar] [CrossRef]
- Yee, R.; Dien Bard, J.; Simner, P.J. The genotype-to-phenotype dilemma: How should laboratories approach discordant susceptibility results? J. Clin. Microbiol. 2021, 59, e00138-20. [Google Scholar] [CrossRef]
- Miller, R.R.; Montoya, V.; Gardy, J.L.; Patrick, D.M.; Tang, P. Metagenomics for pathogen detection in public health. Genome Med. 2013, 5, 81. [Google Scholar] [CrossRef]
- Jensen, H. AVMA US Pet Ownership and Demographics Sourcebook. In Proceedings of the American Veterinary Medical Association, Indianapolis, IN, USA, 21–25 July 2017. [Google Scholar]
- Overgaauw, P.A.; Vinke, C.M.; van Hagen, M.A.; Lipman, L.J. A one health perspective on the human–companion animal relationship with emphasis on zoonotic aspects. Int. J. Environ. Res. Public Health 2020, 17, 3789. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| NCBI RefSeq | BioProject | Collection | Country | Origin |
|---|---|---|---|---|
| ID | ID | Year | ||
| GCF_001281505 | PRJNA256353 | 2013 | USA | Peritoneal abscess |
| GCF_001293125 | PRJNA256353 | 2011 | USA | Abscess |
| GCF_003241315 | PRJNA376580 | 2014 | USA | NICU environment |
| GCF_030826395 | PRJNA872116 | 2019 | USA | Skin |
| GCF_032574585 | PRJNA294605 | 2014 | USA | Infant ICU gut |
| GCF_040117505 | PRJNA1095233 | 2023 | Switzerland | Lung abscess |
| GCF_043427535 | PRJNA987158 | 2021 | USA | Swine farm worker skin |
| GCF_900610365 | PRJEB29478 | 2018 | Switzerland | Breast abscess |
| GCF_905373635 | PRJEB43277 | 2021 | Unknown | Oral cavity |
| GCF_936933995 | PRJEB51076 | 2023 | USA | Skin |
| GCF_943913485 | PRJEB47281 | 2022 | USA | Skin |
| GCF_946223255 | PRJEB47281 | 2022 | Italy | Skin |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tóth, A.G.; Solymosi, N.; Tenk, M.; Káldy, Z.; Németh, T. First Animal Source Metagenome Assembly of Lawsonella clevelandensis from Canine External Otitis. Pathogens 2025, 14, 465. https://doi.org/10.3390/pathogens14050465
Tóth AG, Solymosi N, Tenk M, Káldy Z, Németh T. First Animal Source Metagenome Assembly of Lawsonella clevelandensis from Canine External Otitis. Pathogens. 2025; 14(5):465. https://doi.org/10.3390/pathogens14050465
Chicago/Turabian StyleTóth, Adrienn Gréta, Norbert Solymosi, Miklós Tenk, Zsófia Káldy, and Tibor Németh. 2025. "First Animal Source Metagenome Assembly of Lawsonella clevelandensis from Canine External Otitis" Pathogens 14, no. 5: 465. https://doi.org/10.3390/pathogens14050465
APA StyleTóth, A. G., Solymosi, N., Tenk, M., Káldy, Z., & Németh, T. (2025). First Animal Source Metagenome Assembly of Lawsonella clevelandensis from Canine External Otitis. Pathogens, 14(5), 465. https://doi.org/10.3390/pathogens14050465