
The 16SrXII-P Phytoplasma GOE Is Separated from Other Stolbur Phytoplasmas by Key Genomic Features

Abstract
1. Introduction
2. Materials and Methods
2.1. Genomic Data
2.2. Phylogenetic Comparison
2.3. Ortholog Prediction
2.4. Functional Reconstruction
3. Results
3.1. Genomic Benchmarks of Complete Stolbur Phytoplasma Genomes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 16SrXII-subgroup | 16SrXII-P | 16SrXII-A | 16SrXII-B/-C | |||||
| Taxon | ‘Ca. P. solani’ | ‘Ca. P. australiense’ | ||||||
| Strain | GOE | c1 | c4 | c5 | o3 | PAa | NZSb11 | |
| Chromosome | ||||||||
| Accession | CP155828 | CP103788 | CP103787 | CP103786 | CP103785 | AM422018 | CP002548 | |
| Length (bp) | 704,525 | 751,320 | 751,188 | 824,084 | 973,640 | 879,324 | 959,779 | |
| GC content (%) | 26.17 | 28.37 | 28.37 | 28.07 | 28.58 | 27 | 27 | |
| No. of CDSs (protein coding) | 663 | 719 | 724 | 807 | 1000 | 684 | 1100 | |
| Coding density (genes/kb) | 0.941 | 0.956 | 0.963 | 0.979 | 1.027 | 0.777 | 1.146 | |
| No. of rRNA operons | 2 | 2 | 2 | 2 | 2 | 2 | 2 | |
| No. of tRNAs | 32 | 32 | 32 | 32 | 32 | 35 | 35 | |
| No. of tmRNAs | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| No. of ncRNAs | 2 | 2 | 2 | 2 | 2 | 2 | 2 | |
| Plasmids | ||||||||
| Accession | DQ318777 | |||||||
| Length (bp) | 3635 | |||||||
| No. of CDSs (protein coding) | 4 | |||||||
| Host for reconstruction | Pentastiridius leporinus | Bindweed | Bindweed | Bindweed | Stinging nettle | Cotton | Strawberry | |
| References | [35] | [61] | [62] | [59,63] | ||||
3.2. Phylogenetic Assessment
3.2.1. Average Nucleotide Identity and Sequence Synteny
3.2.2. Marker Gene Analysis
3.3. Functional Comparison and Reconstruction
3.3.1. Pan-Genome Analysis
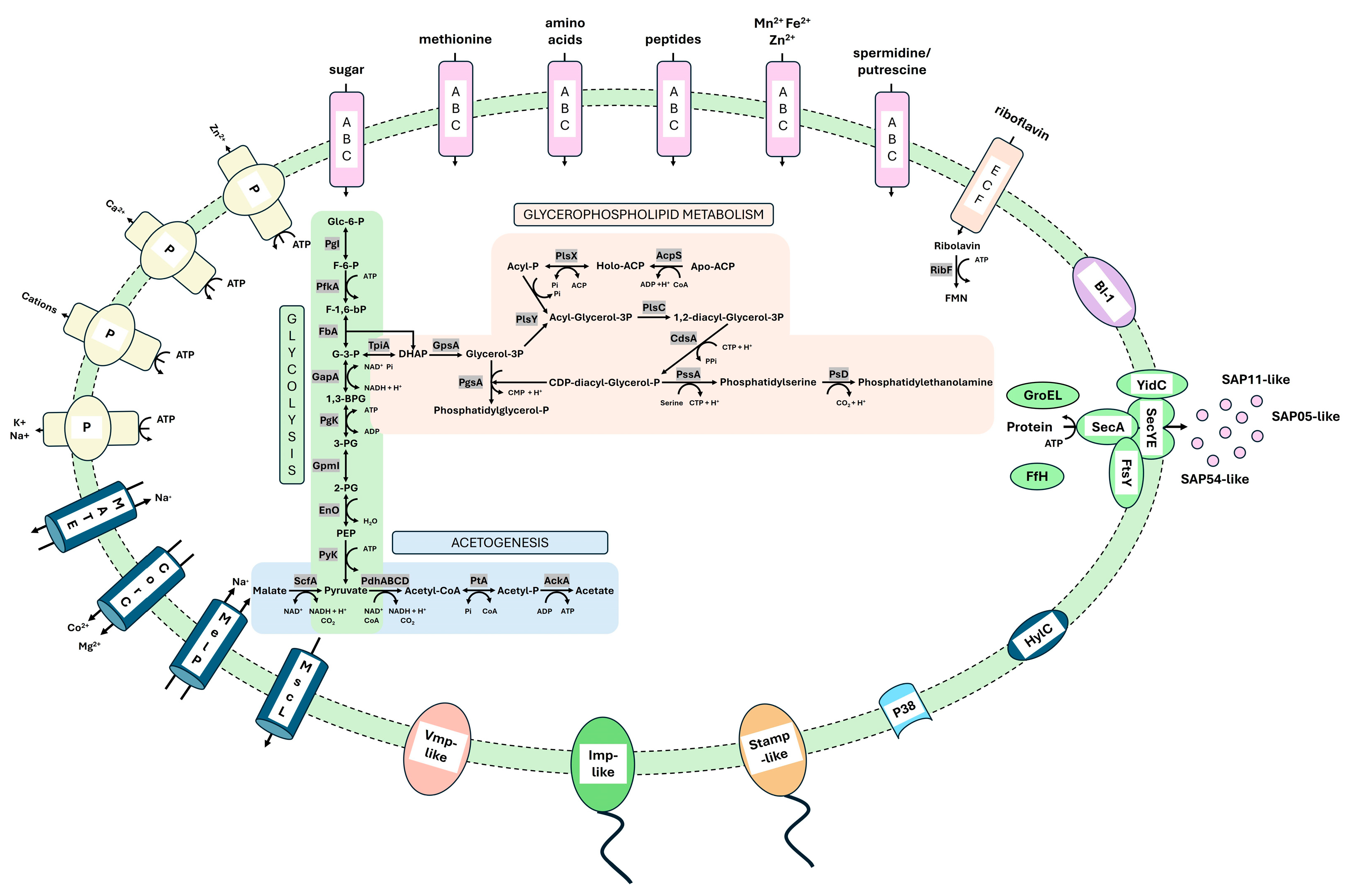
3.3.2. Metabolic Pathways
3.3.3. Transporter
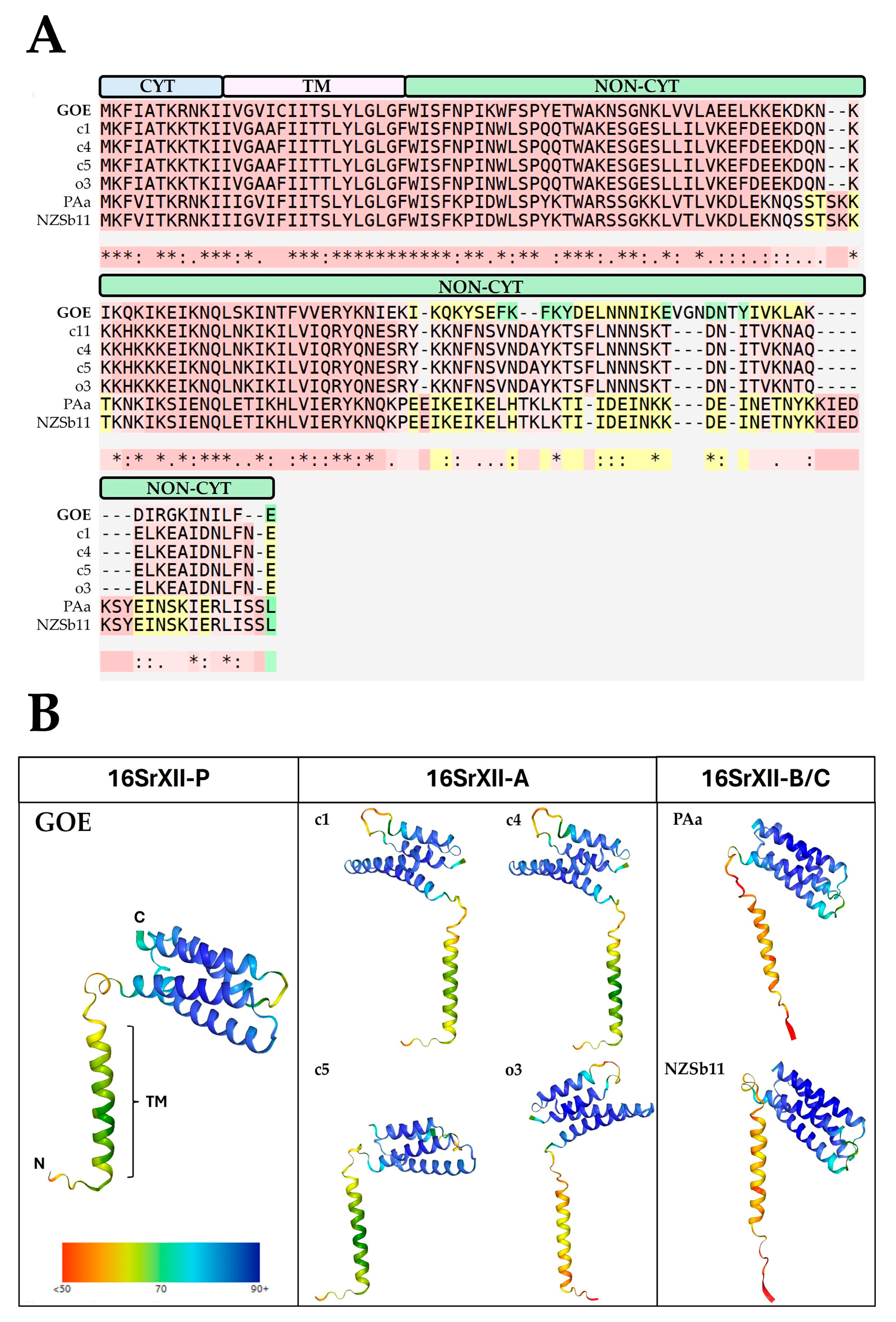
3.3.4. Membrane-Associated Interaction
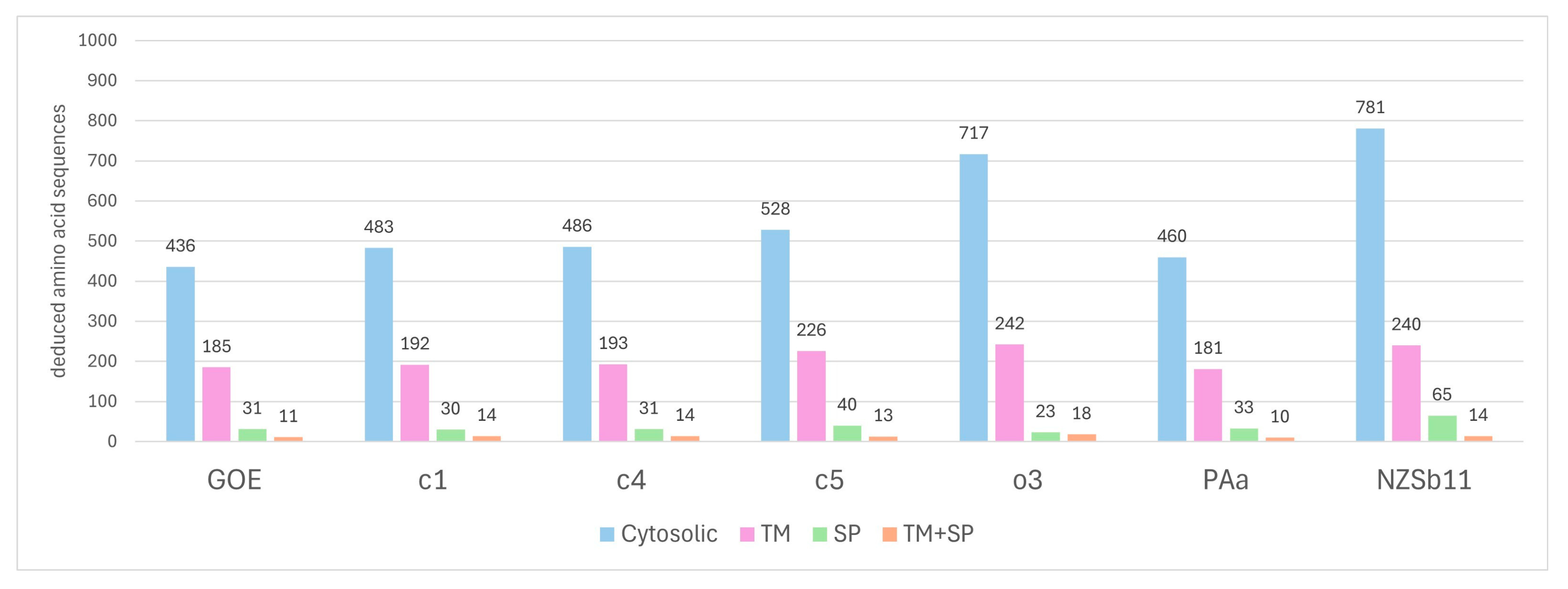
3.3.5. Secretome Analysis
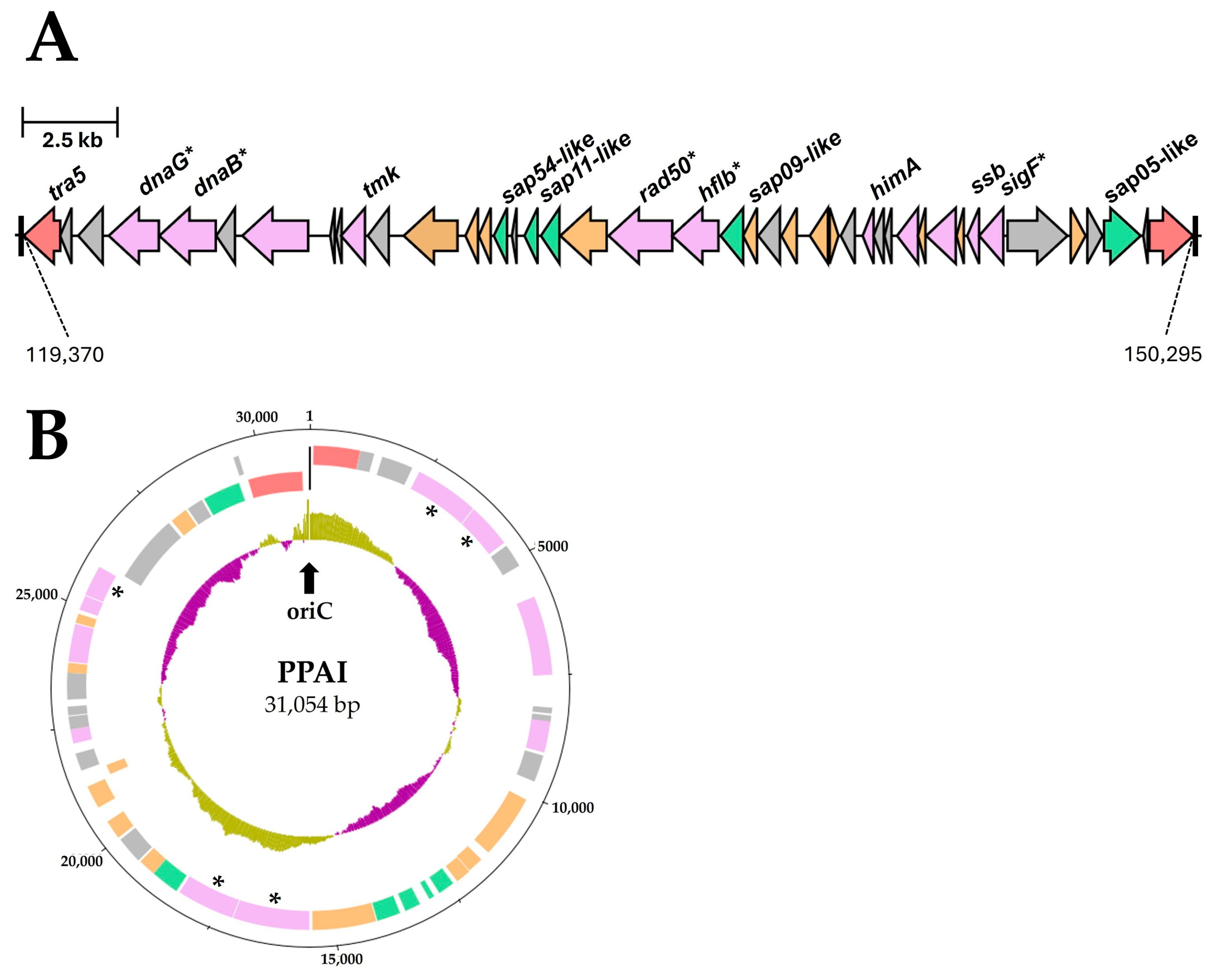
3.3.6. Important Effector Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Richard-Molard, M.; Garessus, S.; Malatesta, G.; Valentin, P.; Fonné, G.; Gerst, M.; Grousson, C. Le syndrôme des “basses richesses”: Investigations au champ et tentatives d’identification de l’agent pathogène et du vecteur. In Proceedings of the 58th Congres de L’Institut International de Recherches Betteravieres, Bruxelles, France, 19 June 1995; pp. 19–22. [Google Scholar]
- Muchembled, C.; Garressus, S.; Ecalle, F.; Boudon-Padieu, E.; Gatineau, F. Low sugar content syndrome. In Proceedings of the Fifth International Conference on Pests in Agriculture, Montpellier, France, 7–9 December 1999; pp. 529–536. [Google Scholar]
- Bressan, A.; Sémétey, O.; Nusillard, B.; Clair, D.; Boudon-Padieu, E. Insect Vectors (Hemiptera: Cixiidae) and Pathogens Associated with the Disease Syndrome “Basses Richesses” of Sugar Beet in France. Plant Dis. 2008, 92, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Gatineau, F.; Larrue, J.; Clair, D.; Lorton, F.; Richard-Molard, M.; Boudon-Padieu, E. A New Natural Planthopper Vector of Stolbur Phytoplasma in the Genus Pentastiridius (Hemiptera: Cixiidae). Eur. J. Plant Pathol. 2001, 107, 263–271. [Google Scholar] [CrossRef]
- Gatineau, F.; Jacob, N.; Vautrin, S.; Larrue, J.; Lherminier, J.; Richard-Molard, M.; Boudon-Padieu, E. Association with the Syndrome “Basses Richesses” of Sugar Beet of a Phytoplasma and a Bacterium-Like Organism Transmitted by a Pentastiridius sp. Phytopathology 2002, 92, 384–392. [Google Scholar] [CrossRef]
- Sémétey, O.; Bressan, A.; Richard-Molard, M.; Boudon-Padieu, E. Monitoring of proteobacteria and phytoplasma in sugar beets naturally or experimentally affected by the disease Syndrome “Basses richesses”. Eur. J. Plant Pathol. 2007, 117, 187–196. [Google Scholar] [CrossRef]
- Behrmann, S.; Schwind, M.; Schieler, M.; Vilcinskas, A.; Martinez, O.; Lee, K.-Z.; Lang, C. Spread of bacterial and virus yellowing diseases of sugar beet in South and Central Germany from 2017–2020. Sugar Ind. 2021, 146, 476–485. [Google Scholar] [CrossRef]
- Bressan, A.; Moral García, F.J.; Boudon-Padieu, E. The prevalence of ‘Candidatus Arsenophonus phytopathogenicus’ infecting the planthopper Pentastiridius leporinus (Hemiptera: Cixiidae) increase nonlinearly with the population abundance in sugar beet fields. Environ. Entomol. 2011, 40, 1345–1352. [Google Scholar] [CrossRef]
- Behrmann, S.C.; Witczak, N.; Lang, C.; Schieler, M.; Dettweiler, A.; Kleinhenz, B.; Schwind, M.; Vilcinskas, A.; Lee, K.-Z. Biology and Rearing of an Emerging Sugar Beet Pest: The Planthopper Pentastiridius leporinus. Insects 2022, 13, 656. [Google Scholar] [CrossRef]
- Pfitzer, R.; Rostás, M.; Häußermann, P.; Häuser, T.; Rinklef, A.; Detring, J.; Schrameyer, K.; Voegele, R.T.; Maier, J.; Varrelmann, M. Effects of succession crops and soil tillage on suppressing the syndrome “basses richesses” vector Pentastiridius leporinus in sugar beet. Pest Manag. Sci. 2024, 80, 3379–3388. [Google Scholar] [CrossRef]
- Schröder, M.; Rissler, D.; Schrameyer, K. Syndrome des“Basses Richesses” (SBR)—Erstmaliges Auftreten an Zuckerrübe in Deutschland. J. Kulturpflanz. 2012, 64, 396–397. [Google Scholar]
- Pfitzer, R.; Schrameyer, K.; Voegele, R.T.; Maier, J.; Lang, C.; Varrelmann, M. Ursachen und Auswirkungen des Auftretens von Syndrome des “Basses Richesses” in deutschen Zuckerrübenanbaugebieten. Sugar Ind. 2020, 145, 234–244. [Google Scholar]
- Mahillon, M.; Groux, R.; Bussereau, F.; Brodard, J.; Debonneville, C.; Demal, S.; Kellenberger, I.; Peter, M.; Steinger, T.; Schumpp, O. Virus Yellows and Syndrome “Basses Richesses” in Western Switzerland: A Dramatic 2020 Season Calls for Urgent Control Measures. Pathogens 2022, 11, 885. [Google Scholar] [CrossRef] [PubMed]
- Behrmann, S.C.; Rinklef, A.; Lang, C.; Vilcinskas, A.; Lee, K.-Z. Potato (Solanum tuberosum) as a New Host for Pentastiridius leporinus (Hemiptera: Cixiidae) and ‘Candidatus Arsenophonus phytopathogenicus’. Insects 2023, 14, 281. [Google Scholar] [CrossRef] [PubMed]
- Duduk, B.; Ćurčić, Ž.; Stepanović, J.; Böhm, J.W.; Kosovac, A.; Rekanović, E.; Kube, M. Prevalence of a ‘Candidatus Phytoplasma solani’-Related Strain Designated as New 16SrXII-P Subgroup over ‘Candidatus Arsenophonus phytopathogenicus’ in Sugar Beet in Eastern Germany. Plant Dis. 2023, 107, 3792–3800. [Google Scholar] [CrossRef] [PubMed]
- Duduk, B.; Stepanović, J.; Fránová, J.; Zwolińska, A.; Rekanović, E.; Stepanović, M.; Vučković, N.; Duduk, N.; Vico, I. Geographical variations, prevalence, and molecular dynamics of fastidious phloem-limited pathogens infecting sugar beet across Central Europe. PLoS ONE 2024, 19, e0306136. [Google Scholar] [CrossRef]
- Lee, I.-M.; Gundersen-Rindal, D.E.; Davis, R.E.; Bartoszyk, I.M. Revised Classification Scheme of Phytoplasmas based on RFLP Analyses of 16S rRNA and Ribosomal Protein Gene Sequences. Int. J. Syst. Evol. Microbiol. 1998, 48, 1153–1169. [Google Scholar] [CrossRef]
- Wei, W.; Lee, I.-M.; Davis, R.E.; Suo, X.; Zhao, Y. Automated RFLP pattern comparison and similarity coefficient calculation for rapid delineation of new and distinct phytoplasma 16Sr subgroup lineages. Int. J. Syst. Evol. Microbiol. 2008, 58, 2368–2377. [Google Scholar] [CrossRef]
- Zhao, Y.; Wei, W.; Lee, I.-M.; Shao, J.; Suo, X.; Davis, R.E. Construction of an interactive online phytoplasma classification tool, iPhyClassifier, and its application in analysis of the peach X-disease phytoplasma group (16SrIII). Int. J. Syst. Evol. Microbiol. 2009, 59, 2582–2593. [Google Scholar] [CrossRef]
- Jović, J.; Ember, I.; Mitrović, M.; Cvrković, T.; Krstić, O.; Krnjajić, S.; Acs, Z.; Kölber, M.; Toševski, I. Molecular detection of potato stolbur phytoplasma in Serbia. Bull. Insectology 2011, 64, 83–84. [Google Scholar]
- Favali, M.A.; Musetti, R.; Fossati, F.; Vighi, C. Association of stolbur phytoplasmas with diseased tomatoes in Italy. EPPO Bull. 2000, 30, 347–350. [Google Scholar] [CrossRef]
- Duduk, B.; Bertaccini, A. Corn with Symptoms of Reddening: New Host of Stolbur Phytoplasma. Plant Dis. 2006, 90, 1313–1319. [Google Scholar] [CrossRef]
- Jović, J.; Cvrković, T.; Mitrović, M.; Krnjajić, S.; Redinbaugh, M.G.; Pratt, R.C.; Gingery, R.E.; Hogenhout, S.A.; Toševski, I. Roles of stolbur phytoplasma and Reptalus panzeri (Cixiidae, Auchenorrhyncha) in the epidemiology of Maize redness in Serbia. Eur. J. Plant Pathol. 2007, 118, 85–89. [Google Scholar] [CrossRef]
- Jović, J.; Cvrković, T.; Mitrović, M.; Krnjajić, S.; Petrović, A.; Redinbaugh, M.G.; Pratt, R.C.; Hogenhout, S.A.; Tosevski, I. Stolbur phytoplasma transmission to maize by Reptalus panzeri and the disease cycle of maize redness in Serbia. Phytopathology 2009, 99, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Boudon-Padieu, E.; Cousin, M.T. Yellow decline of Lavandula hybrida Rev and L. vera DC. Int. J. Trop. Plant Dis. 1999, 17, 1–34. [Google Scholar]
- Sémétey, O.; Gaudin, J.; Danet, J.-L.; Salar, P.; Theil, S.; Fontaine, M.; Krausz, M.; Chaisse, E.; Eveillard, S.; Verdin, E.; et al. Lavender Decline in France Is Associated with Chronic Infection by Lavender-Specific Strains of ‘Candidatus Phytoplasma solani’. Appl. Environ. Microbiol. 2018, 84, e01507-18. [Google Scholar] [CrossRef]
- Davis, R.E.; Dally, E.L.; Gundersen, D.E.; Lee, I.M.; Habili, N. ‘Candidatus Phytoplasma australiense’, a new phytoplasma taxon associated with Australian grapevine yellows. Int. J. Syst. Evol. Microbiol. 1997, 47, 262–269. [Google Scholar] [CrossRef]
- Sawayanagi, T.; Horikoshi, N.; Kanehira, T.; Shinohara, M.; Bertaccini, A.; Cousin, M.T.; Hiruki, C.; Namba, S. ‘Candidatus Phytoplasma japonicum’, a new phytoplasma taxon associated with Japanese Hydrangea phyllody. Int. J. Syst. Evol. Microbiol. 1999, 49 Pt 3, 1275–1285. [Google Scholar] [CrossRef]
- Valiunas, D.; Staniulis, J.; Davis, R.E. ‘Candidatus Phytoplasma fragariae’, a novel phytoplasma taxon discovered in yellows diseased strawberry, Fragaria x ananassa. Int. J. Syst. Evol. Microbiol. 2006, 56, 277–281. [Google Scholar] [CrossRef]
- Ćurčić, Ž.; Stepanović, J.; Zübert, C.; Taški-Ajduković, K.; Kosovac, A.; Rekanović, E.; Kube, M.; Duduk, B. Rubbery Taproot Disease of Sugar Beet in Serbia Associated with ‘Candidatus Phytoplasma solani’. Plant Dis. 2021, 105, 255–263. [Google Scholar] [CrossRef]
- Eini, O.; Shoaei, Z.; Varrelmann, M. Molecular detection and multilocus sequence analysis of ‘Candidatus Phytoplasma solani’-related strains infecting potato and sugar beet plants in Southern Germany. Res. Sq. 2024; submitted. [Google Scholar] [CrossRef]
- Toth, R.; Ilic, A.-M.; Huettel, B.; Duduk, B.; Kube, M. Divergence within the Taxon ‘Candidatus Phytoplasma asteris’ Confirmed by Comparative Genome Analysis of Carrot Strains. Microorganisms 2024, 12, 1016. [Google Scholar] [CrossRef]
- Kube, M.; Mitrovic, J.; Duduk, B.; Rabus, R.; Seemüller, E. Current view on phytoplasma genomes and encoded metabolism. Sci. World J. 2012, 2012, 185942. [Google Scholar] [CrossRef]
- Bertaccini, A.; Oshima, K.; Maejima, K.; Namba, S. Phytoplasma Effectors and Pathogenicity Factors. In Phytoplasmas: Plant Pathogenic Bacteria—III; Springer: Singapore, 2019; pp. 17–34. ISBN 978-981-13-9632-8. [Google Scholar]
- Toth, R.; Huettel, B.; Eini, O.; Varrelmann, M.; Kube, M. The complete genome sequence of the stolbur pathogen ‘Candidatus Phytoplasma solani’ from Pentastiridius leporinus. Microbiol. Resour. Announc. 2024, 14, e0064024. [Google Scholar] [CrossRef] [PubMed]
- Carver, T.; Harris, S.R.; Berriman, M.; Parkhill, J.; McQuillan, J.A. Artemis: An integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 2012, 28, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Gundersen, D.E.; Lee, I.-M. Ultrasensitive detection of phytoplasmas by nested-PCR assays using two universal primer pairs. Phytopathol. Mediterr. 1996, 35, 144–151. [Google Scholar]
- Schneider, B.; Gibb, K.S. Sequence and RFLP analysis of the elongation factor Tu gene used in differentiation and classification of phytoplasmas. Microbiology 1997, 143 Pt 10, 3381–3389. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Zhao, Y.; Wei, W.; Lee, I.-M.; Shao, J.; Suo, X.; Davis, R.E. The iPhyClassifier, an interactive online tool for phytoplasma classification and taxonomic assignment. Phytoplasma 2013, 938, 329–338. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Mathelier, A. Intervene: A tool for intersection and visualization of multiple gene or genomic region sets. BMC Bioinform. 2017, 18, 287. [Google Scholar] [CrossRef] [PubMed]
- Zdobnov, E.M.; Apweiler, R. InterProScan—An integration platform for the signature-recognition methods in InterPro. Bioinform. 2001, 17, 847–848. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Ong, W.K.; Paley, S.; Subhraveti, P.; Karp, P.D. The MetaCyc database of metabolic pathways and enzymes—A 2019 update. Nucleic Acids Res. 2020, 48, D445–D453. [Google Scholar] [CrossRef]
- Tully, J.G.; Whitcomb, R.F.; Rose, D.L.; Bové, J.M.; Carle, P.; Somerson, N.L.; Williamson, D.L.; Eden-Green, S. Acholeplasma brassicae sp. nov. and Acholeplasma palmae sp. nov., two non-sterol-requiring Mollicutes from plant surfaces. Int. J. Syst. Evol. Microbiol. 1994, 44, 680–684. [Google Scholar] [CrossRef]
- Käll, L.; Krogh, A.; Sonnhammer, E.L.L. Advantages of combined transmembrane topology and signal peptide prediction—The Phobius web server. Nucleic Acids Res. 2007, 35, W429–W432. [Google Scholar] [CrossRef]
- Teufel, F.; Almagro Armenteros, J.J.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 2022, 40, 1023–1025. [Google Scholar] [CrossRef]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. Int. J. Syst. Evol. Microbiol. 2000, 302, 205–217. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Tang, H. ISEScan: Automated identification of insertion sequence elements in prokaryotic genomes. Bioinformatics 2017, 33, 3340–3347. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, C.L.M.; Chooi, Y.-H. clinker & clustermap.js: Automatic generation of gene cluster comparison figures. Bioinformatics 2021, 37, 2473–2475. [Google Scholar] [CrossRef] [PubMed]
- Zübert, C.; Ilic, A.-M.; Duduk, B.; Kube, M. The Genome Reduction Excludes the Ribosomal Rescue System in Acholeplasmataceae. Microb. Physiol. 2022, 32, 45–56. [Google Scholar] [CrossRef]
- Liefting, L.W.; Andersen, M.T.; Lough, T.J.; Beever, R.E. Comparative analysis of the plasmids from two isolates of ‘Candidatus Phytoplasma australiense’. Plasmid 2006, 56, 138–144. [Google Scholar] [CrossRef]
- Tran-Nguyen, L.T.T.; Gibb, K.S. Extrachromosomal DNA isolated from tomato big bud and ‘Candidatus Phytoplasma australiense’ phytoplasma strains. Plasmid 2006, 56, 153–166. [Google Scholar] [CrossRef]
- Carminati, G. ‘Candidatus Phytoplasma solani’: From Insect Transmission to Complete Genome Sequencing of Several Strains Inducing Different Symptoms in Tomato. Ph.D. Thesis, University of Udine, Udine, Italy, 13 March 2023. [Google Scholar]
- Tran-Nguyen, L.T.T.; Kube, M.; Schneider, B.; Reinhardt, R.; Gibb, K.S. Comparative genome analysis of ‘Candidatus Phytoplasma australiense ‘(subgroup tuf-Australia I; rp-A) and ‘Ca. Phytoplasma asteris’ strains OY-M and AY-WB. J. Bacteriol. 2008, 190, 3979–3991. [Google Scholar] [CrossRef]
- Andersen, M.T.; Liefting, L.W.; Havukkala, I.; Beever, R.E. Comparison of the complete genome sequence of two closely related isolates of ‘Candidatus Phytoplasma australiense’ reveals genome plasticity. BMC Genom. 2013, 14, 529. [Google Scholar] [CrossRef]
- Bertaccini, A.; Arocha-Rosete, Y.; Contaldo, N.; Duduk, B.; Fiore, N.; Montano, H.G.; Kube, M.; Kuo, C.-H.; Martini, M.; Oshima, K.; et al. Revision of the ‘Candidatus Phytoplasma’ species description guidelines. Int. J. Syst. Evol. Microbiol. 2022, 72, 5353. [Google Scholar] [CrossRef]
- Seemüller, E.; Schneider, B.; Mäurer, R.; Ahrens, U.; Daire, X.; Kison, H.; Lorenz, K.H.; Firrao, G.; Avinent, L.; Sears, B.B. Phylogenetic classification of phytopathogenic mollicutes by sequence analysis of 16S ribosomal DNA. Int. J. Syst. Bacteriol. 1994, 44, 440–446. [Google Scholar] [CrossRef]
- Liefting, L.W.; Andersen, M.T.; Beever, R.E.; Gardner, R.C.; Forster, R.L. Sequence heterogeneity in the two 16S rRNA genes of Phormium yellow leaf phytoplasma. Appl. Environ. Microbiol. 1996, 62, 3133–3139. [Google Scholar] [CrossRef] [PubMed]
- Jomantiene, R.; Davis, R.E.; Valiunas, D.; Alminaite, A. New Group 16SrIII Phytoplasma Lineages in Lithuania Exhibit rRNA Interoperon Sequence Heterogeneity. Eur. J. Plant Pathol. 2002, 108, 507–517. [Google Scholar] [CrossRef]
- Saigo, M.; Golic, A.; Alvarez, C.E.; Andreo, C.S.; Hogenhout, S.A.; Mussi, M.A.; Drincovich, M.F. Metabolic regulation of phytoplasma malic enzyme and phosphotransacetylase supports the use of malate as an energy source in these plant pathogens. Microbiology 2014, 160, 2794–2806. [Google Scholar] [CrossRef] [PubMed]
- Kakizawa, S.; Oshima, K.; Namba, S. Diversity and functional importance of phytoplasma membrane proteins. Trends Microbiol. 2006, 14, 254–256. [Google Scholar] [CrossRef]
- Fabre, A.; Danet, J.-L.; Foissac, X. The stolbur phytoplasma antigenic membrane protein gene stamp is submitted to diversifying positive selection. Gene 2011, 472, 37–41. [Google Scholar] [CrossRef]
- Boonrod, K.; Munteanu, B.; Jarausch, B.; Jarausch, W.; Krczal, G. An immunodominant membrane protein (Imp) of ‘Candidatus Phytoplasma mali’ binds to plant actin. Mol. Plant Microbe Interact. 2012, 25, 889–895. [Google Scholar] [CrossRef]
- Konnerth, A.; Krczal, G.; Boonrod, K. Immunodominant membrane proteins of phytoplasmas. Microbiology 2016, 162, 1267–1273. [Google Scholar] [CrossRef]
- Neriya, Y.; Maejima, K.; Nijo, T.; Tomomitsu, T.; Yusa, A.; Himeno, M.; Netsu, O.; Hamamoto, H.; Oshima, K.; Namba, S. Onion yellow phytoplasma P38 protein plays a role in adhesion to the hosts. FEMS Microbiol. Lett. 2014, 361, 115–122. [Google Scholar] [CrossRef]
- Cimerman, A.; Pacifico, D.; Salar, P.; Marzachì, C.; Foissac, X. Striking diversity of vmp1, a variable gene encoding a putative membrane protein of the stolbur phytoplasma. Appl. Environ. Microbiol. 2009, 75, 2951–2957. [Google Scholar] [CrossRef]
- Hückelhoven, R.; Dechert, C.; Kogel, K.-H. Overexpression of barley BAX inhibitor 1 induces breakdown of mlo-mediated penetration resistance to Blumeria graminis. Proc. Natl. Acad. Sci. USA 2003, 100, 5555–5560. [Google Scholar] [CrossRef]
- Quaglino, F.; Kube, M.; Jawhari, M.; Abou-Jawdah, Y.; Siewert, C.; Choueiri, E.; Sobh, H.; Casati, P.; Tedeschi, R.; Lova, M.M.; et al. ‘Candidatus Phytoplasma phoenicium’ associated with almond witches’-broom disease: From draft genome to genetic diversity among strain populations. BMC Microbiol. 2015, 15, 148. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Ren, Z.; Zhao, W.; Li, W. ‘Candidatus Phytoplasma ziziphi’ encodes non-classically secreted proteins that suppress hypersensitive cell death response in Nicotiana benthamiana. Phytopathol. Res. 2023, 5, 11. [Google Scholar] [CrossRef]
- Passera, A.; Zhao, Y.; Murolo, S.; Pierro, R.; Arsov, E.; Mori, N.; Moussa, A.; Silletti, M.R.; Casati, P.; Panattoni, A.; et al. Multilocus Genotyping Reveals New Molecular Markers for Differentiating Distinct Genetic Lineages among ‘Candidatus Phytoplasma solani’ Strains Associated with Grapevine Bois Noir. Pathogens 2020, 9, 970. [Google Scholar] [CrossRef] [PubMed]
- Kakizawa, S.; Oshima, K.; Nishigawa, H.; Jung, H.-Y.; Wei, W.; Suzuki, S.; Tanaka, M.; Miyata, S.-I.; Ugaki, M.; Namba, S. Secretion of immunodominant membrane protein from onion yellows phytoplasma through the Sec protein-translocation system in Escherichia coli. Microbiology 2004, 150, 135–142. [Google Scholar] [CrossRef]
- Koch, H.-G.; Moser, M.; Müller, M. Signal recognition particle-dependent protein targeting, universal to all kingdoms of life. Rev. Physiol. Biochem. Pharmacol. 2003, 146, 55–94. [Google Scholar] [CrossRef]
- Bai, X.; Zhang, J.; Ewing, A.; Miller, S.A.; Jancso Radek, A.; Shevchenko, D.V.; Tsukerman, K.; Walunas, T.; Lapidus, A.; Campbell, J.W.; et al. Living with Genome Instability: The Adaptation of Phytoplasmas to Diverse Environments of Their Insect and Plant Hosts. J. Bacteriol. 2006, 188, 3682–3696. [Google Scholar] [CrossRef]
- Sugio, A.; Kingdom, H.N.; MacLean, A.M.; Grieve, V.M.; Hogenhout, S.A. Phytoplasma protein effector SAP11 enhances insect vector reproduction by manipulating plant development and defense hormone biosynthesis. Proc. Natl. Acad. Sci. USA 2011, 108, E1254–E1263. [Google Scholar] [CrossRef]
- Drcelic, M.; Skiljaica, A.; Polak, B.; Bauer, N.; Seruga Music, M. ‘Candidatus Phytoplasma solani’ Predicted Effector SAP11-like Alters Morphology of Transformed Arabidopsis Plants and Interacts with AtTCP2 and AtTCP4 Plant Transcription Factors. Pathogens 2024, 13, 893. [Google Scholar] [CrossRef]
- MacLean, A.M.; Sugio, A.; Makarova, O.V.; Findlay, K.C.; Grieve, V.M.; Tóth, R.; Nicolaisen, M.; Hogenhout, S.A. Phytoplasma effector SAP54 induces indeterminate leaf-like flower development in Arabidopsis plants. Plant Physiol. 2011, 157, 831–841. [Google Scholar] [CrossRef]
- Huang, W.; MacLean, A.M.; Sugio, A.; Maqbool, A.; Busscher, M.; Cho, S.-T.; Kamoun, S.; Kuo, C.-H.; Immink, R.G.H.; Hogenhout, S.A. Parasitic modulation of host development by ubiquitin-independent protein degradation. Cell 2021, 184, 5201–5214.e12. [Google Scholar] [CrossRef]
- Music, M.S.; Samarzija, I.; Hogenhout, S.A.; Haryono, M.; Cho, S.-T.; Kuo, C.-H. The genome of ‘Candidatus Phytoplasma solani’ strain SA-1 is highly dynamic and prone to adopting foreign sequences. Syst. Appl. Microbiol. 2019, 42, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Mahillon, J.; Chandler, M. Insertion sequences. Microbiol. Mol. Biol. Rev. 1998, 62, 725–774. [Google Scholar] [CrossRef]
- Toruño, T.Y.; Musić, M.S.; Simi, S.; Nicolaisen, M.; Hogenhout, S.A. Phytoplasma PMU1 exists as linear chromosomal and circular extrachromosomal elements and has enhanced expression in insect vectors compared with plant hosts. Mol. Microbiol. 2010, 77, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Kube, M.; Schneider, B.; Kuhl, H.; Dandekar, T.; Heitmann, K.; Migdoll, A.M.; Reinhardt, R.; Seemüller, E. The linear chromosome of the plant-pathogenic mycoplasma ‘Candidatus Phytoplasma mali’. BMC Genom. 2008, 9, 306. [Google Scholar] [CrossRef] [PubMed]
- Böhm, J.W.; Duckeck, D.; Duduk, B.; Schneider, B.; Kube, M. Genome Comparison of ‘Candidatus Phytoplasma rubi’ with Genomes of Other 16SrV Phytoplasmas Highlights Special Group Features. Appl. Microbiol. 2023, 3, 1083–1100. [Google Scholar] [CrossRef]
- Dong, H.; Beer, S.V. Riboflavin induces disease resistance in plants by activating a novel signal transduction pathway. Phytopathology 2000, 90, 801–811. [Google Scholar] [CrossRef]
- Sugio, A.; MacLean, A.M.; Kingdom, H.N.; Grieve, V.M.; Manimekalai, R.; Hogenhout, S.A. Diverse targets of phytoplasma effectors: From plant development to defense against insects. Annu. Rev. Phytopathol. 2011, 49, 175–195. [Google Scholar] [CrossRef]
- Cho, S.-T.; Kung, H.-J.; Huang, W.; Hogenhout, S.A.; Kuo, C.-H. Species Boundaries and Molecular Markers for the Classification of 16SrI Phytoplasmas Inferred by Genome Analysis. Front. Microbiol. 2020, 11, 1531. [Google Scholar] [CrossRef]
- MacLean, A.M.; Orlovskis, Z.; Kowitwanich, K.; Zdziarska, A.M.; Angenent, G.C.; Immink, R.G.H.; Hogenhout, S.A. Phytoplasma effector SAP54 hijacks plant reproduction by degrading MADS-box proteins and promotes insect colonization in a RAD23-dependent manner. PLoS Biol. 2014, 12, e1001835. [Google Scholar] [CrossRef]
- Orlovskis, Z.; Singh, A.; Kliot, A.; Huang, W.; Hogenhout, S.A. Molecular Matchmakers: Phytoplasma Effector SAP54 Targets MADS-Box Factor SVP to Enhance Attraction of Fecund Female Vectors by Modulating Leaf Responses to Male Presence. bioRxiv 2024. [Google Scholar] [CrossRef]
- Tokuda, R.; Iwabuchi, N.; Kitazawa, Y.; Nijo, T.; Suzuki, M.; Maejima, K.; Oshima, K.; Namba, S.; Yamaji, Y. Potential mobile units drive the horizontal transfer of phytoplasma effector phyllogen genes. Front. Genet. 2023, 14, 1132432. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.-C.; Chen, L.-L.; Lo, W.-S.; Lin, C.-P.; Kuo, C.-H. Comparative analysis of the peanut witches’-broom phytoplasma genome reveals horizontal transfer of potential mobile units and effectors. PLoS ONE 2013, 8, e62770. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-T.; Cho, S.-T.; Lin, Y.-C.; Tan, C.-M.; Chiu, Y.-C.; Yang, J.-Y.; Kuo, C.-H. Comparative Genome Analysis of ‘Candidatus Phytoplasma luffae’ Reveals the Influential Roles of Potential Mobile Units in Phytoplasma Evolution. Front. Microbiol. 2022, 13, 773608. [Google Scholar] [CrossRef] [PubMed]
- Kohl, S.; Bock, R. Transposition of a bacterial insertion sequence in chloroplasts. Plant J. 2009, 58, 423–436. [Google Scholar] [CrossRef]

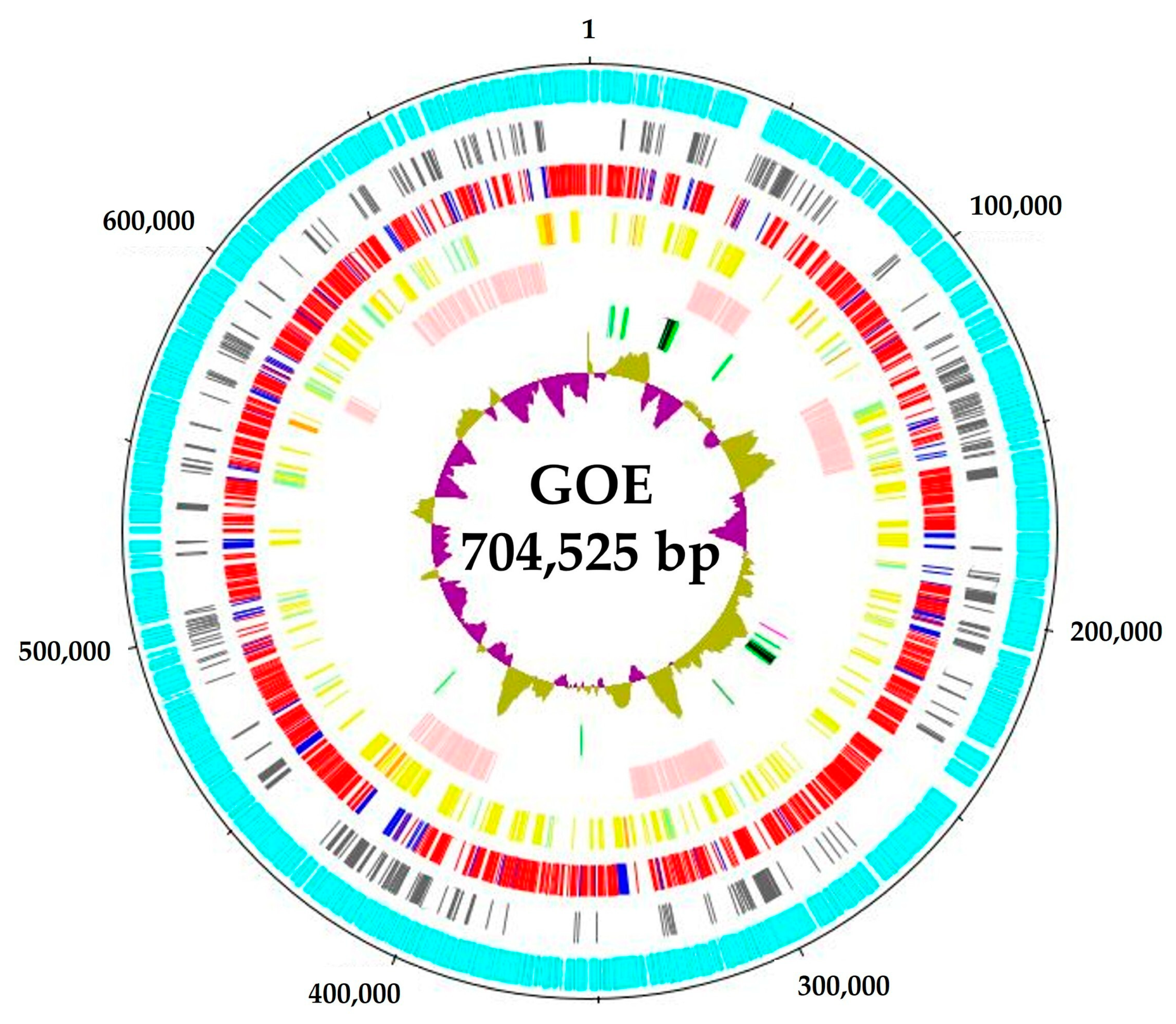
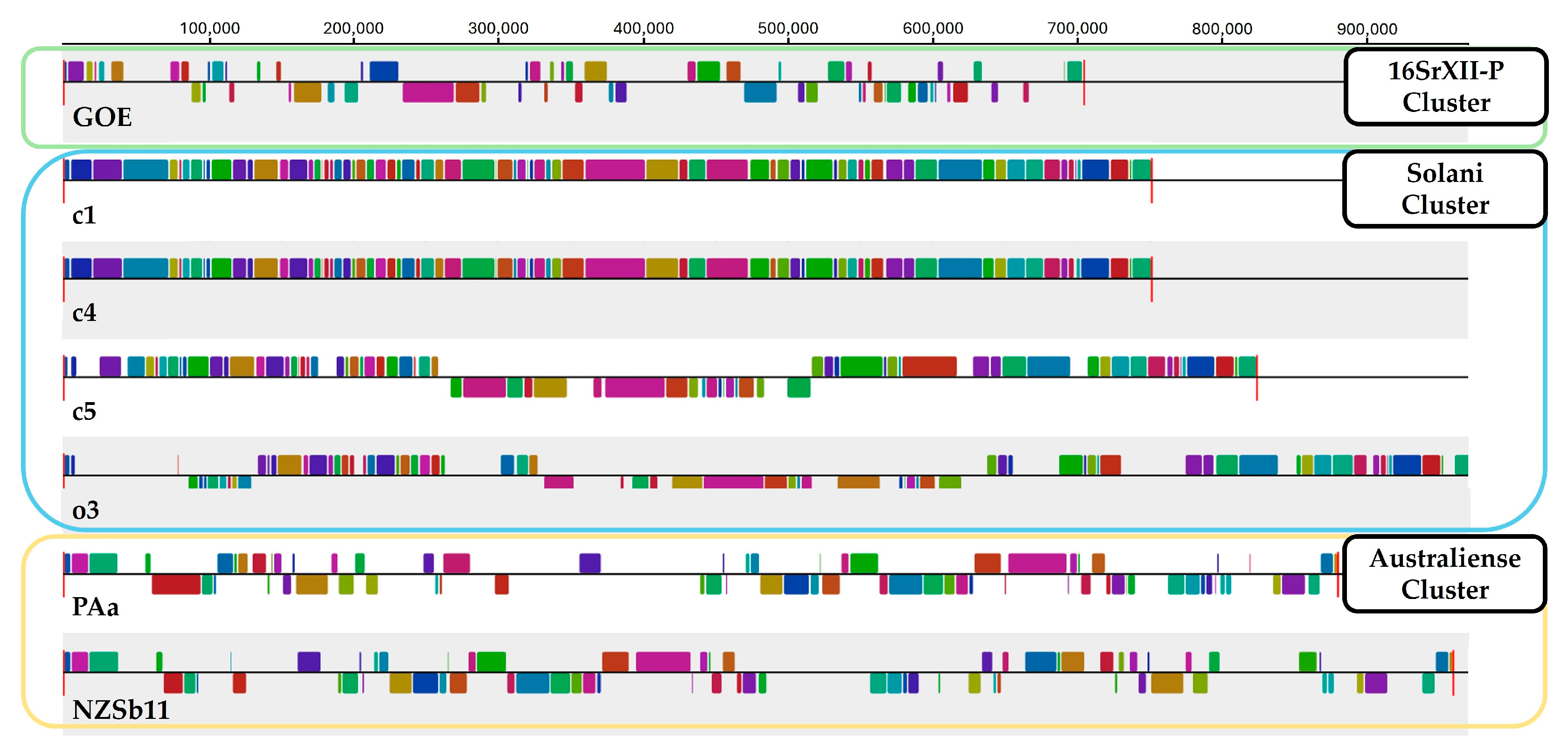
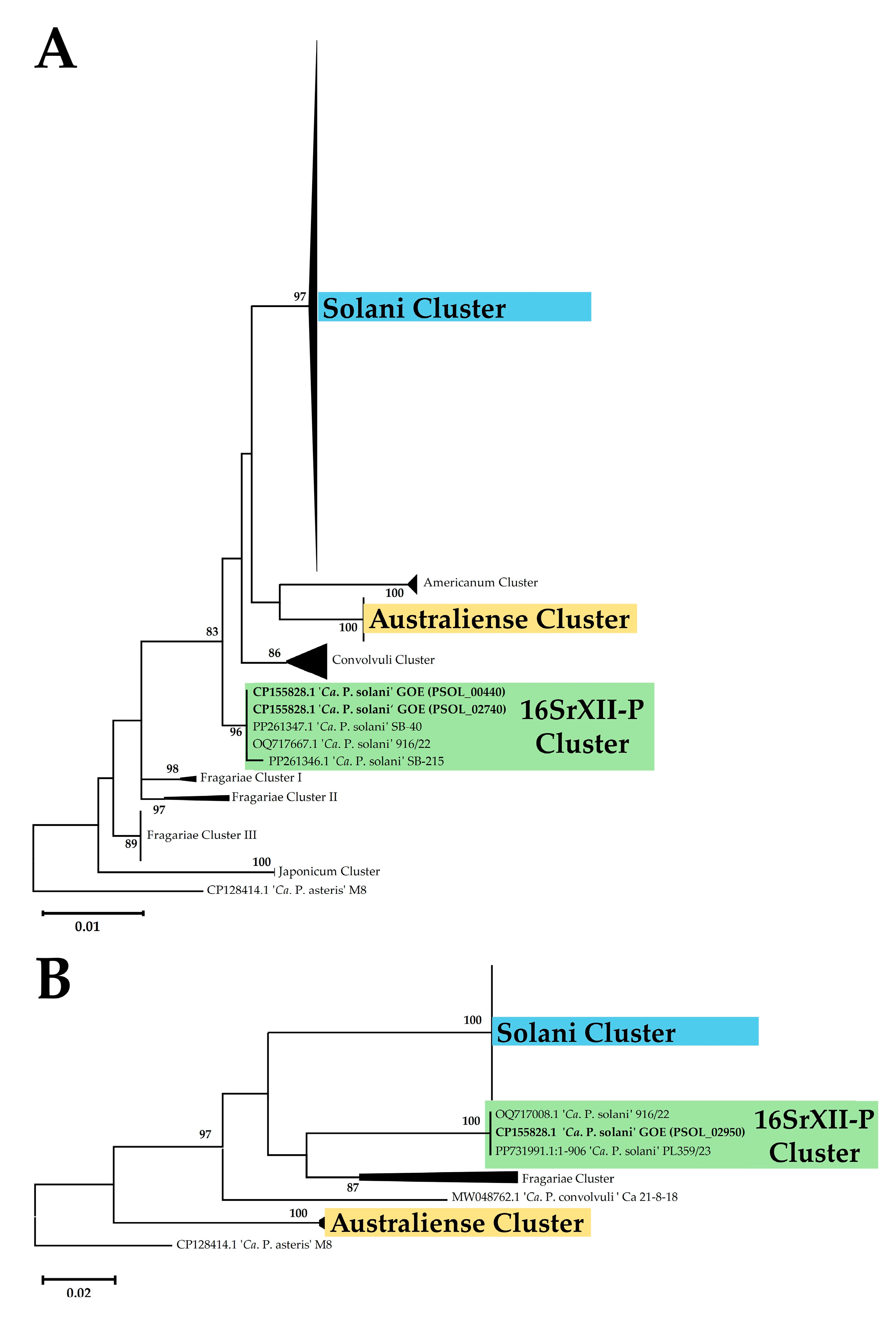


| Cluster | 16SrXII-P | Solani | Australiense | ||||
|---|---|---|---|---|---|---|---|
| Strain | GOE | c1 | c4 | c5 | o3 | PAa | NZSb11 |
| GOE | - | 83.04 | 83.05 | 82.87 | 82.85 | 82.00 | 82.23 |
| c1 | 83.04 | - | 99.99 | 99.15 | 98.42 | 79.71 | 79.99 |
| c4 | 83.05 | 99.98 | - | 99.15 | 98.40 | 79.73 | 80.04 |
| c5 | 82.87 | 99.15 | 99.15 | - | 98.53 | 79.94 | 80.02 |
| o3 | 82.85 | 98.42 | 98.40 | 98.53 | - | 80.12 | 79.57 |
| PAa | 82.00 | 79.71 | 79.73 | 79.94 | 80.12 | - | 98.64 |
| NZSb11 | 82.23 | 79.99 | 80.04 | 80.02 | 79.57 | 98.64 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toth, R.; Huettel, B.; Varrelmann, M.; Kube, M. The 16SrXII-P Phytoplasma GOE Is Separated from Other Stolbur Phytoplasmas by Key Genomic Features. Pathogens 2025, 14, 180. https://doi.org/10.3390/pathogens14020180
Toth R, Huettel B, Varrelmann M, Kube M. The 16SrXII-P Phytoplasma GOE Is Separated from Other Stolbur Phytoplasmas by Key Genomic Features. Pathogens. 2025; 14(2):180. https://doi.org/10.3390/pathogens14020180
Chicago/Turabian StyleToth, Rafael, Bruno Huettel, Mark Varrelmann, and Michael Kube. 2025. "The 16SrXII-P Phytoplasma GOE Is Separated from Other Stolbur Phytoplasmas by Key Genomic Features" Pathogens 14, no. 2: 180. https://doi.org/10.3390/pathogens14020180
APA StyleToth, R., Huettel, B., Varrelmann, M., & Kube, M. (2025). The 16SrXII-P Phytoplasma GOE Is Separated from Other Stolbur Phytoplasmas by Key Genomic Features. Pathogens, 14(2), 180. https://doi.org/10.3390/pathogens14020180