
The Intricate Interplay between APOBEC3 Proteins and DNA Tumour Viruses


Abstract
1. Introduction
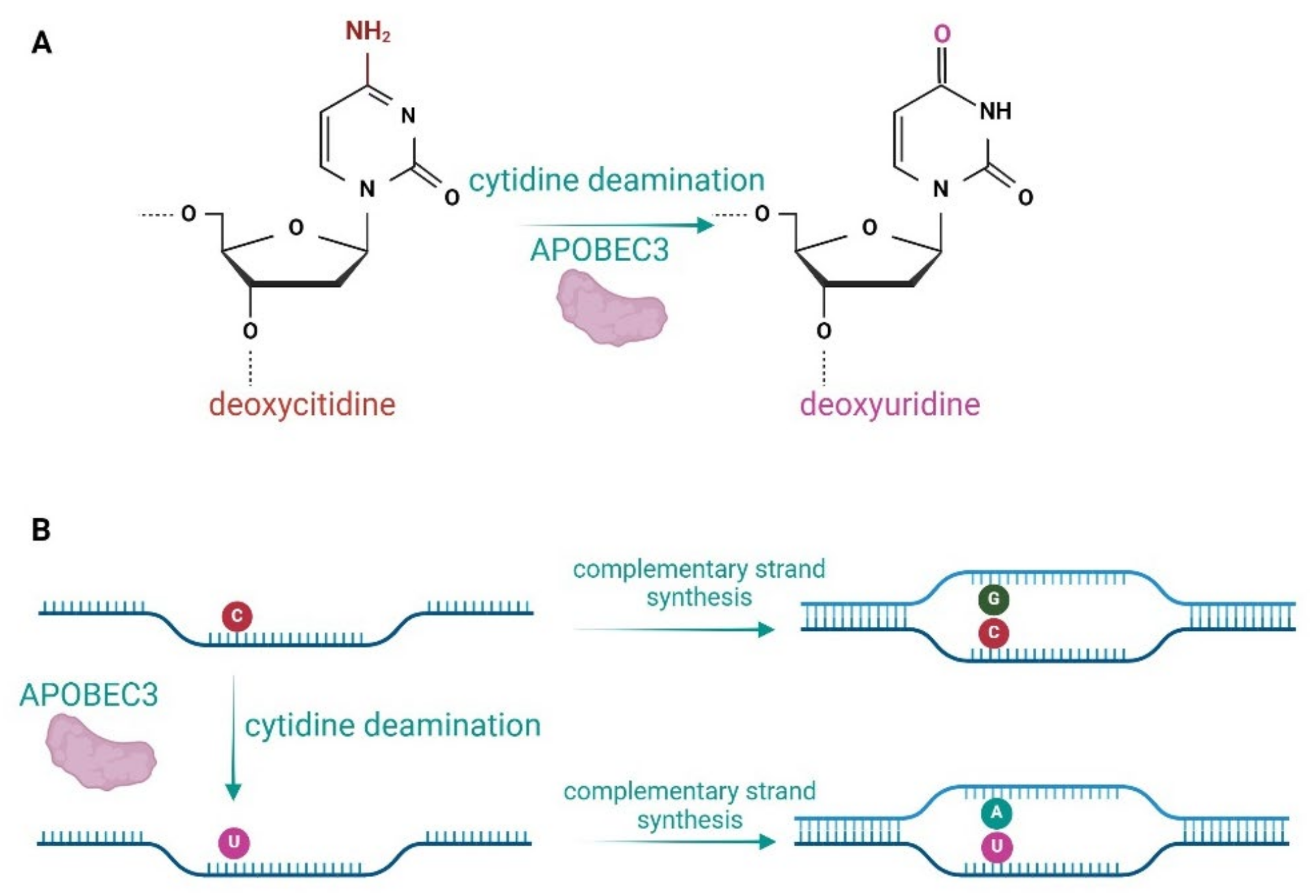
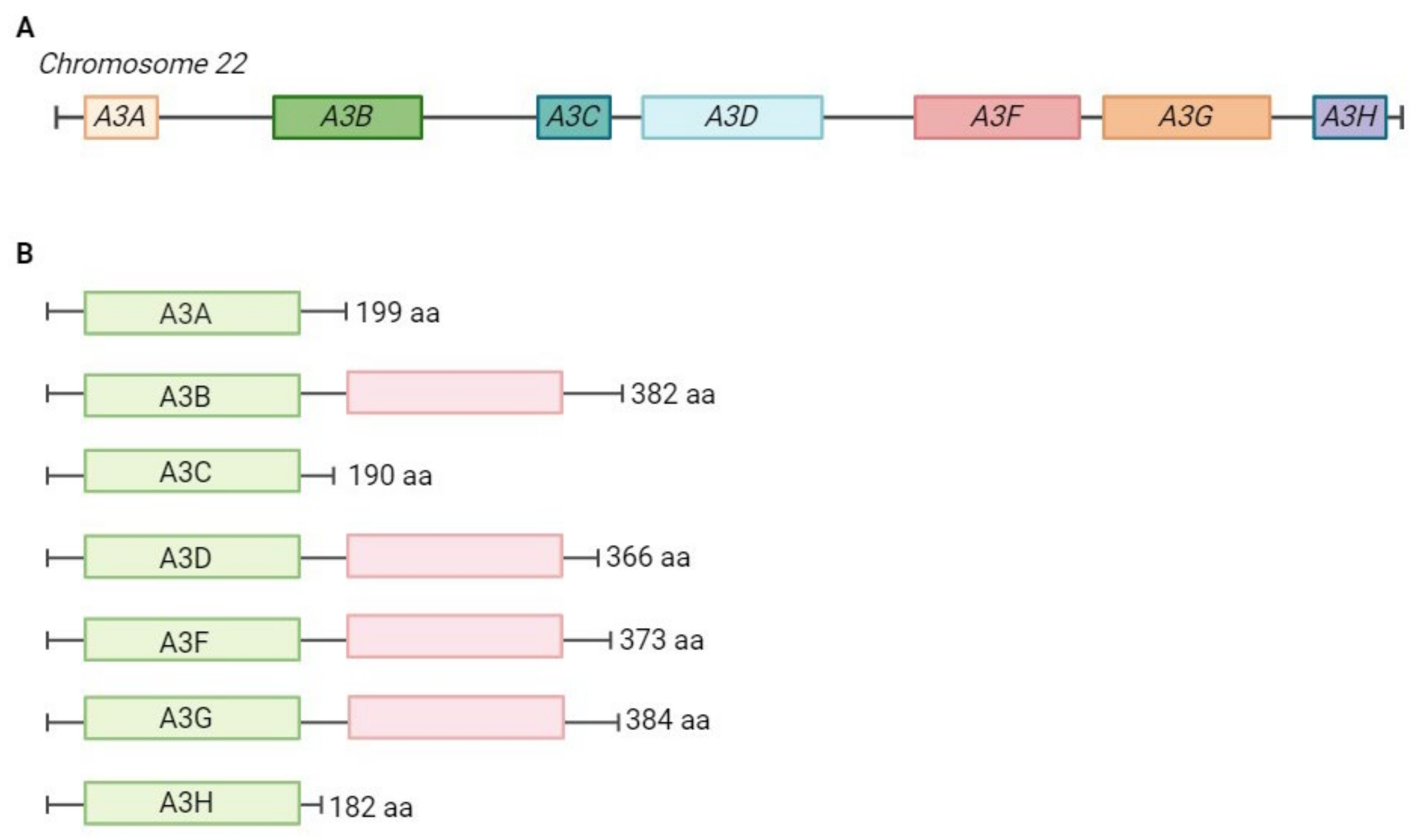
2. APOBEC Proteins and Their Role in Viral Inhibition and Genome Editing
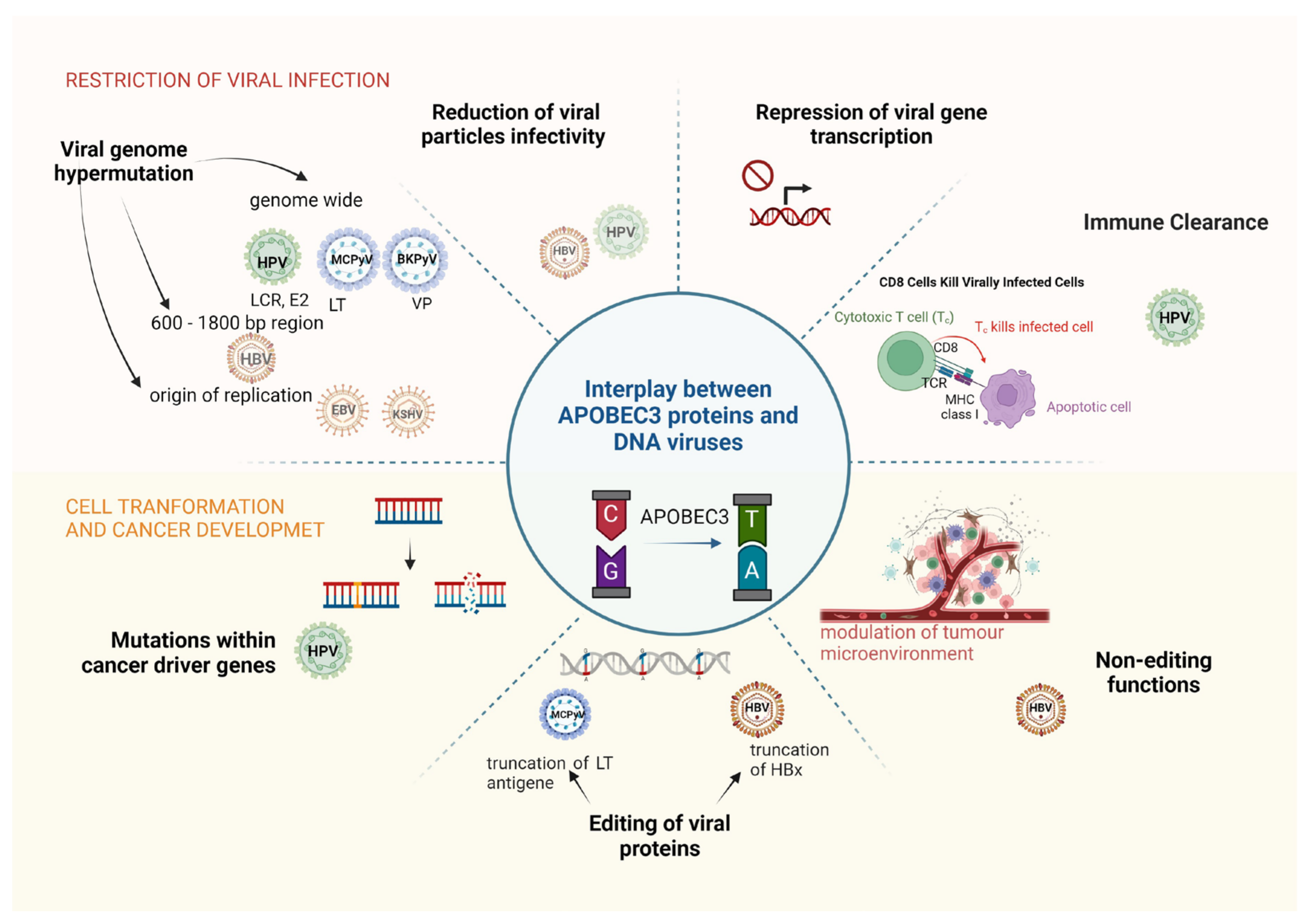
3. Restriction of DNA Viruses by APOBEC Proteins
3.1. Hepatitis B Virus
3.2. Human Papillomaviruses
3.3. Human Polyomaviruses
3.4. Gamma Herpesviruses
4. How APOBEC3 Proteins Contribute to the Oncogenicity of DNA Viruses?
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zheng, Y.-H.; Lovsin, N.; Peterlin, B.M. Newly Identified Host Factors Modulate HIV Replication. Immunol. Lett. 2005, 97, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.-L.; Greene, W.C. The APOBEC3 Cytidine Deaminases: An Innate Defensive Network Opposing Exogenous Retroviruses and Endogenous Retroelements. Annu. Rev. Immunol. 2008, 26, 317–353. [Google Scholar] [CrossRef] [PubMed]
- Stavrou, S.; Ross, S.R. APOBEC3 Proteins in Viral Immunity. J. Immunol. 2015, 195, 4565–4570. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N. The Repertoire of Mutational Signatures in Human Cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Goubau, D.; Deddouche, S.; e Sousa, C.R. Cytosolic Sensing of Viruses. Immunity 2013, 38, 855–869. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Yu, Y.; Liu, B.; Luo, K.; Kong, W.; Mao, P.; Yu, X.-F. Induction of APOBEC3G Ubiquitination and Degradation by an HIV-1 Vif-Cul5-SCF Complex. Science 2003, 302, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.Z.; Yockteng-Melgar, J.; Jarvis, M.C.; Malik-Soni, N.; Borozan, I.; Carpenter, M.A.; McCann, J.L.; Ebrahimi, D.; Shaban, N.M.; Marcon, E. Epstein–Barr Virus BORF2 Inhibits Cellular APOBEC3B to Preserve Viral Genome Integrity. Nat. Microbiol. 2019, 4, 78–88. [Google Scholar] [CrossRef]
- Poulain, F.; Lejeune, N.; Willemart, K.; Gillet, N.A. Footprint of the Host Restriction Factors APOBEC3 on the Genome of Human Viruses. PLoS Pathog. 2020, 16, e1008718. [Google Scholar] [CrossRef]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a Human Gene That Inhibits HIV-1 Infection and Is Suppressed by the Viral Vif Protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef]
- Krump, N.A.; You, J. Molecular Mechanisms of Viral Oncogenesis in Humans. Nat. Rev. Microbiol. 2018, 16, 684–698. [Google Scholar] [CrossRef]
- Schiller, J.T.; Lowy, D.R. An Introduction to Virus Infections and Human Cancer. Recent Results Cancer Res. 2021, 217, 1–11. [Google Scholar]
- Liu, X.; Marmorstein, R. When Viral Oncoprotein Meets Tumor Suppressor: A Structural View. Genes Dev. 2006, 20, 2332–2337. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Dudley, J.P. APOBECs and Virus Restriction. Virology 2015, 479–480, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Hakata, Y.; Miyazawa, M. Deaminase-Independent Mode of Antiretroviral Action in Human and Mouse APOBEC3 Proteins. Microorganisms 2020, 8, 1976. [Google Scholar] [CrossRef] [PubMed]
- Knisbacher, B.A.; Gerber, D.; Levanon, E.Y. DNA Editing by APOBECs: A Genomic Preserver and Transformer. Trends Genet. 2016, 32, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Patnaik, S.K.; Thomas Taggart, R.; Kannisto, E.D.; Enriquez, S.M.; Gollnick, P.; Baysal, B.E. APOBEC3A Cytidine Deaminase Induces RNA Editing in Monocytes and Macrophages. Nat. Commun. 2015, 6, 6881. [Google Scholar] [CrossRef]
- Ratcliff, J.; Simmonds, P. Potential APOBEC-Mediated RNA Editing of the Genomes of SARS-CoV-2 and Other Coronaviruses and Its Impact on Their Longer Term Evolution. Virology 2021, 556, 62–72. [Google Scholar] [CrossRef]
- Kim, K.; Shi, A.B.; Kelley, K.; Chen, X.S. Unraveling the Enzyme-Substrate Properties for APOBEC3A-Mediated RNA Editing. J. Mol. Biol. 2023, 435, 168198. [Google Scholar] [CrossRef]
- Honjo, T. Does AID Need Another Aid? Nat. Immunol. 2002, 3, 800–801. [Google Scholar] [CrossRef] [PubMed]
- Conticello, S.G. The AID/APOBEC Family of Nucleic Acid Mutators. Genome Biol. 2008, 9, 229. [Google Scholar] [CrossRef]
- Severi, F.; Chicca, A.; Conticello, S.G. Analysis of Reptilian APOBEC1 Suggests That RNA Editing May Not Be Its Ancestral Function. Mol. Biol. Evol. 2011, 28, 1125–1129. [Google Scholar] [CrossRef] [PubMed]
- Milewska, A.; Kindler, E.; Vkovski, P.; Zeglen, S.; Ochman, M.; Thiel, V.; Rajfur, Z.; Pyrc, K. APOBEC3-Mediated Restriction of RNA Virus Replication. Sci. Rep. 2018, 8, 5960. [Google Scholar] [CrossRef] [PubMed]
- Jarmuz, A.; Chester, A.; Bayliss, J.; Gisbourne, J.; Dunham, I.; Scott, J.; Navaratnam, N. An Anthropoid-Specific Locus of Orphan C to U RNA-Editing Enzymes on Chromosome 22. Genomics 2002, 79, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Bishop, K.N.; Sheehy, A.M.; Craig, H.M.; Petersen-Mahrt, S.K.; Watt, I.N.; Neuberger, M.S.; Malim, M.H. DNA Deamination Mediates Innate Immunity to Retroviral Infection. Cell 2003, 113, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Okeoma, C.M.; Lovsin, N.; Peterlin, B.M.; Ross, S.R. APOBEC3 Inhibits Mouse Mammary Tumour Virus Replication in Vivo. Nature 2007, 445, 927–930. [Google Scholar] [CrossRef] [PubMed]
- Bogerd, H.P.; Wiegand, H.L.; Hulme, A.E.; Garcia-Perez, J.L.; O’Shea, K.S.; Moran, J.V.; Cullen, B.R. Cellular Inhibitors of Long Interspersed Element 1 and Alu Retrotransposition. Proc. Natl. Acad. Sci. USA 2006, 103, 8780–8785. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.; Fenton, T. APOBEC3 Genes: Retroviral Restriction Factors to Cancer Drivers. Trends Mol. Med. 2015, 21, 274–284. [Google Scholar] [CrossRef]
- Bhutani, N.; Burns, D.M.; Blau, H.M. DNA Demethylation Dynamics. Cell 2011, 146, 866–872. [Google Scholar] [CrossRef]
- Cervantes-Gracia, K.; Gramalla-Schmitz, A.; Weischedel, J.; Chahwan, R. APOBECs Orchestrate Genomic and Epigenomic Editing across Health and Disease. Trends Genet. 2021, 37, 1028–1043. [Google Scholar] [CrossRef]
- Cen, S.; Guo, F.; Niu, M.; Saadatmand, J.; Deflassieux, J.; Kleiman, L. The Interaction between HIV-1 Gag and APOBEC3G. J. Biol. Chem. 2004, 279, 33177–33184. [Google Scholar] [CrossRef]
- Zheng, Y.-H.; Irwin, D.; Kurosu, T.; Tokunaga, K.; Sata, T.; Peterlin, B.M. Human APOBEC3F Is Another Host Factor That Blocks Human Immunodeficiency Virus Type 1 Replication. J. Virol. 2004, 78, 6073–6076. [Google Scholar] [CrossRef]
- Aguiar, R.S.; Lovsin, N.; Tanuri, A.; Peterlin, B.M. Vpr.A3A Chimera Inhibits HIV Replication. J. Biol. Chem. 2008, 283, 2518–2525. [Google Scholar] [CrossRef]
- Mangeat, B.; Turelli, P.; Caron, G.; Friedli, M.; Perrin, L.; Trono, D. Broad Antiretroviral Defence by Human APOBEC3G through Lethal Editing of Nascent Reverse Transcripts. Nature 2003, 424, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Doehle, B.P.; Schäfer, A.; Wiegand, H.L.; Bogerd, H.P.; Cullen, B.R. Differential Sensitivity of Murine Leukemia Virus to APOBEC3-Mediated Inhibition Is Governed by Virion Exclusion. J. Virol. 2005, 79, 8201–8207. [Google Scholar] [CrossRef] [PubMed]
- Löchelt, M.; Romen, F.; Bastone, P.; Muckenfuss, H.; Kirchner, N.; Kim, Y.-B.; Truyen, U.; Rösler, U.; Battenberg, M.; Saib, A. The Antiretroviral Activity of APOBEC3 Is Inhibited by the Foamy Virus Accessory Bet Protein. Proc. Natl. Acad. Sci. USA 2005, 102, 7982–7987. [Google Scholar] [CrossRef]
- Jaguva Vasudevan, A.A.; Becker, D.; Luedde, T.; Gohlke, H.; Münk, C. Foamy Viruses, Bet, and APOBEC3 Restriction. Viruses 2021, 13, 504. [Google Scholar] [CrossRef]
- Peng, Z.; Zhao, Z.; Li, Y.; Wang, Y.; Hao, L.; Fan, B.; Li, Y.; Wang, Y.; Shan, Y.; Han, Y. Host Apolipoprotein B Messenger RNA-editing Enzyme Catalytic Polypeptide-like 3G Is an Innate Defensive Factor and Drug Target against Hepatitis C Virus. Hepatology 2011, 53, 1080–1089. [Google Scholar] [CrossRef]
- Narvaiza, I.; Linfesty, D.C.; Greener, B.N.; Hakata, Y.; Pintel, D.J.; Logue, E.; Landau, N.R.; Weitzman, M.D. Deaminase-Independent Inhibition of Parvoviruses by the APOBEC3A Cytidine Deaminase. PLoS Pathog. 2009, 5, e1000439. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Huang, J.; Zhang, C.; Huang, S.; Nunnari, G.; Wang, F.; Tong, X.; Gao, L.; Nikisher, K.; Zhang, H. Alpha Interferon Potently Enhances the Anti-Human Immunodeficiency Virus Type 1 Activity of APOBEC3G in Resting Primary CD4 T Cells. J. Virol. 2006, 80, 7645–7657. [Google Scholar] [CrossRef]
- Turelli, P.; Mangeat, B.; Jost, S.; Vianin, S.; Trono, D. Inhibition of Hepatitis B Virus Replication by APOBEC3G. Science 2004, 303, 1829. [Google Scholar] [CrossRef]
- Vartanian, J.-P.; Guétard, D.; Henry, M.; Wain-Hobson, S. Evidence for Editing of Human Papillomavirus DNA by APOBEC3 in Benign and Precancerous Lesions. Science 2008, 320, 230–233. [Google Scholar] [CrossRef]
- Nakata, Y.; Ode, H.; Kubota, M.; Kasahara, T.; Matsuoka, K.; Sugimoto, A.; Imahashi, M.; Yokomaku, Y.; Iwatani, Y. Cellular APOBEC3A Deaminase Drives Mutations in the SARS-CoV-2 Genome. Nucleic Acids Res. 2023, 51, 783–795. [Google Scholar] [CrossRef]
- O’Toole, Á.; Neher, R.A.; Ndodo, N.; Borges, V.; Gannon, B.; Gomes, J.P.; Groves, N.; King, D.J.; Maloney, D.; Lemey, P. APOBEC3 Deaminase Editing in Mpox Virus as Evidence for Sustained Human Transmission since at Least 2016. Science 2023, 382, 595–600. [Google Scholar] [CrossRef]
- Albin, J.S.; Harris, R.S. Interactions of Host APOBEC3 Restriction Factors with HIV-1 in Vivo: Implications for Therapeutics. Expert Rev. Mol. Med. 2010, 12, e4. [Google Scholar] [CrossRef]
- Carmi, S.; Church, G.M.; Levanon, E.Y. Large-Scale DNA Editing of Retrotransposons Accelerates Mammalian Genome Evolution. Nat. Commun. 2011, 2, 519. [Google Scholar] [CrossRef] [PubMed]
- Modenini, G.; Abondio, P.; Boattini, A. The Coevolution between APOBEC3 and Retrotransposons in Primates. Mob. DNA 2022, 13, 27. [Google Scholar] [CrossRef]
- Esnault, C.; Heidmann, O.; Delebecque, F.; Dewannieux, M.; Ribet, D.; Hance, A.J.; Heidmann, T.; Schwartz, O. APOBEC3G Cytidine Deaminase Inhibits Retrotransposition of Endogenous Retroviruses. Nature 2005, 433, 430–433. [Google Scholar] [CrossRef] [PubMed]
- Holtz, C.M.; Sadler, H.A.; Mansky, L.M. APOBEC3G Cytosine Deamination Hotspots Are Defined by Both Sequence Context and Single-Stranded DNA Secondary Structure. Nucleic Acids Res. 2013, 41, 6139–6148. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lindič, N.; Budič, M.; Petan, T.; Knisbacher, B.A.; Levanon, E.Y.; Lovšin, N. Differential Inhibition of LINE1 and LINE2 Retrotransposition by Vertebrate AID/APOBEC Proteins. Retrovirology 2013, 10, 156. [Google Scholar] [CrossRef]
- Holmes, R.K.; Malim, M.H.; Bishop, K.N. APOBEC-Mediated Viral Restriction: Not Simply Editing? Trends Biochem. Sci. 2007, 32, 118–128. [Google Scholar] [CrossRef]
- Feng, Y.; Goubran, M.H.; Follack, T.B.; Chelico, L. Deamination-Independent Restriction of LINE-1 Retrotransposition by APOBEC3H. Sci. Rep. 2017, 7, 10881. [Google Scholar] [CrossRef] [PubMed]
- Lovsin, N.; Peterlin, B.M. APOBEC3 Proteins Inhibit LINE-1 Retrotransposition in the Absence of ORF1p Binding. Ann. N. Y. Acad. Sci. 2009, 1178, 268–275. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, X.; Cao, Y.; Yang, Z. Roles of APOBEC3 in Hepatitis B Virus (HBV) Infection and Hepatocarcinogenesis. Bioengineered 2021, 12, 2074–2086. [Google Scholar] [CrossRef]
- Beggel, B.; Münk, C.; Däumer, M.; Hauck, K.; Häussinger, D.; Lengauer, T.; Erhardt, A. Full Genome Ultra-deep Pyrosequencing Associates G-to-A Hypermutation of the Hepatitis B Virus Genome with the Natural Progression of Hepatitis B. J. Viral Hepat. 2013, 20, 882–889. [Google Scholar] [CrossRef]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-Induced Hepatocellular Carcinoma. J. Hepatol. 2016, 64, S84–S101. [Google Scholar] [CrossRef]
- Tang, H.; Oishi, N.; Kaneko, S.; Murakami, S. Molecular Functions and Biological Roles of Hepatitis B Virus x Protein. Cancer Sci. 2006, 97, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Tsukuda, S.; Watashi, K. Hepatitis B Virus Biology and Life Cycle. Antivir. Res. 2020, 182, 104925. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Eggerman, T.L.; Bocharov, A.V.; Baranova, I.N.; Vishnyakova, T.G.; Patterson, A.P. APOBEC3-Induced Mutation of the Hepatitis Virus B DNA Genome Occurs during Its Viral RNA Reverse Transcription into (−)-DNA. J. Biol. Chem. 2021, 297, 100889. [Google Scholar] [CrossRef]
- Vartanian, J.-P.; Henry, M.; Marchio, A.; Suspène, R.; Aynaud, M.-M.; Guétard, D.; Cervantes-Gonzalez, M.; Battiston, C.; Mazzaferro, V.; Pineau, P. Massive APOBEC3 Editing of Hepatitis B Viral DNA in Cirrhosis. PLoS Pathog. 2010, 6, e1000928. [Google Scholar] [CrossRef]
- Suspène, R.; Guétard, D.; Henry, M.; Sommer, P.; Wain-Hobson, S.; Vartanian, J.-P. Extensive Editing of Both Hepatitis B Virus DNA Strands by APOBEC3 Cytidine Deaminases in Vitro and in Vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 8321–8326. [Google Scholar] [CrossRef]
- Ren, F.; Li, W.; Zhao, S.; Wang, L.; Wang, Q.; Li, M.; Xiang, A.; Guo, Y. A3G-induced Mutations Show a Low Prevalence and Exhibit Plus-strand Regional Distribution in Hepatitis B Virus DNA from Patients with Non-hepatocellular Carcinoma (HCC) and HCC. J. Med. Virol. 2021, 93, 3672–3678. [Google Scholar] [CrossRef]
- Bonvin, M.; Achermann, F.; Greeve, I.; Stroka, D.; Keogh, A.; Inderbitzin, D.; Candinas, D.; Sommer, P.; Wain-Hobson, S.; Vartanian, J. Interferon-inducible Expression of APOBEC3 Editing Enzymes in Human Hepatocytes and Inhibition of Hepatitis B Virus Replication. Hepatology 2006, 43, 1364–1374. [Google Scholar] [CrossRef]
- Henry, M.; Guétard, D.; Suspène, R.; Rusniok, C.; Wain-Hobson, S.; Vartanian, J.-P. Genetic Editing of HBV DNA by Monodomain Human APOBEC3 Cytidine Deaminases and the Recombinant Nature of APOBEC3G. PLoS ONE 2009, 4, e4277. [Google Scholar] [CrossRef]
- Köck, J.; Blum, H.E. Hypermutation of Hepatitis B Virus Genomes by APOBEC3G, APOBEC3C and APOBEC3H. J. Gen. Virol. 2008, 89, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Baumert, T.F.; Rösler, C.; Malim, M.H.; von Weizsäcker, F. Hepatitis B Virus DNA Is Subject to Extensive Editing by the Human Deaminase APOBEC3C. Hepatology 2007, 46, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzl, M.F.; Koppensteiner, H.; Makowska, Z.; Volz, T. Specific and Nonhepatotoxic Degradation of Nuclear Hepatitis B Virus CccDNA. Science 2014, 343, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, M.; Krug, L.T.; MacCarthy, T. Mutational Pressure by Host APOBEC3s More Strongly Affects Genes Expressed Early in the Lytic Phase of Herpes Simplex Virus-1 (HSV-1) and Human Polyomavirus (HPyV) Infection. PLoS Pathog. 2021, 17, e1009560. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xia, Y.; Han, M.; Chen, G.; Zhang, D.; Thasler, W.E.; Protzer, U.; Ning, Q. IFN-α-Mediated Base Excision Repair Pathway Correlates with Antiviral Response against Hepatitis B Virus Infection. Sci. Rep. 2017, 7, 12715. [Google Scholar] [CrossRef]
- Bouzidi, M.S.; Caval, V.; Suspène, R.; Hallez, C.; Pineau, P.; Wain-Hobson, S.; Vartanian, J.-P. APOBEC3DE Antagonizes Hepatitis B Virus Restriction Factors APOBEC3F and APOBEC3G. J. Mol. Biol. 2016, 428, 3514–3528. [Google Scholar] [CrossRef] [PubMed]
- Rösler, C.; Köck, J.; Kann, M.; Malim, M.H.; Blum, H.E.; Baumert, T.F.; von Weizsäcker, F. APOBEC-mediated Interference with Hepadnavirus Production. Hepatology 2005, 42, 301–309. [Google Scholar] [CrossRef]
- Abe, H.; Ochi, H.; Maekawa, T.; Hatakeyama, T.; Tsuge, M.; Kitamura, S.; Kimura, T.; Miki, D.; Mitsui, F.; Hiraga, N. Effects of Structural Variations of APOBEC3A and APOBEC3B Genes in Chronic Hepatitis B Virus Infection. Hepatol. Res. 2009, 39, 1159–1168. [Google Scholar] [CrossRef]
- Kanagaraj, A.; Sakamoto, N.; Que, L.; Li, Y.; Mohiuddin, M.; Koura, M.; Wakae, K.; Kurachi, M.; Muramatsu, M.; Kitamura, K. Different Antiviral Activities of Natural APOBEC3C, APOBEC3G, and APOBEC3H Variants against Hepatitis B Virus. Biochem. Biophys. Res. Commun. 2019, 518, 26–31. [Google Scholar] [CrossRef]
- Ezzikouri, S.; Kitab, B.; Rebbani, K.; Marchio, A.; Wain-Hobson, S.; Dejean, A.; Vartanian, J.; Pineau, P.; Benjelloun, S. Polymorphic APOBEC 3 Modulates Chronic Hepatitis B in M Oroccan Population. J. Viral Hepat. 2013, 20, 678–686. [Google Scholar] [CrossRef]
- Lei, Y.-C.; Tian, Y.-J.; Ding, H.-H.; Wang, B.-J.; Yang, Y.; Hao, Y.-H.; Zhao, X.-P.; Lu, M.-J.; Gong, F.-L.; Yang, D.-L. N-Terminal and C-Terminal Cytosine Deaminase Domain of APOBEC3G Inhibit Hepatitis B Virus Replication. World J. Gastroenterol. WJG 2006, 12, 7488. [Google Scholar] [CrossRef]
- Migita, K.; Abiru, S.; Ohtani, M.; Jiuchi, Y.; Maeda, Y.; Bae, S.K.; Bekki, S.; Hashimoto, S.; Yesmembetov, K.; Nagaoka, S. HLA-DP Gene Polymorphisms and Hepatitis B Infection in the Japanese Population. Transl. Res. 2012, 160, 443–444. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, X.; Tian, C.; Wang, T.; Sarkis, P.T.N.; Fang, Y.; Zheng, S.; Yu, X.; Xu, R. Cytidine Deaminase APOBEC3B Interacts with Heterogeneous Nuclear Ribonucleoprotein K and Suppresses Hepatitis B Virus Expression. Cell Microbiol. 2008, 10, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.H.; Gummuluru, S.; Hu, J. Deamination-Independent Inhibition of Hepatitis B Virus Reverse Transcription by APOBEC3G. J. Virol. 2007, 81, 4465–4472. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Wang, Z.; Chowdhury, S.; Simadu, M.; Koura, M.; Muramatsu, M. Uracil DNA Glycosylase Counteracts APOBEC3G-Induced Hypermutation of Hepatitis B Viral Genomes: Excision Repair of Covalently Closed Circular DNA. PLoS Pathog. 2013, 9, e1003361. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Fukano, K.; Que, L.; Li, Y.; Wakae, K.; Muramatsu, M. Activities of Endogenous APOBEC3s and Uracil-DNA-Glycosylase Affect the Hypermutation Frequency of Hepatitis B Virus CccDNA. J. Gen. Virol. 2022, 103, 1732. [Google Scholar] [CrossRef] [PubMed]
- Broniarczyk, J.; Massimi, P.; Pim, D.; Bergant Marušič, M.; Myers, M.P.; Garcea, R.L.; Banks, L. Phosphorylation of Human Papillomavirus Type 16 L2 Contributes to Efficient Virus Infectious Entry. J. Virol. 2019, 93, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Bergant Marušič, M.; Ozbun, M.A.; Campos, S.K.; Myers, M.P.; Banks, L. Human Papillomavirus L2 Facilitates Viral Escape from Late Endosomes via Sorting Nexin 17. Traffic 2012, 13, 455–467. [Google Scholar] [CrossRef]
- Siddiqa, A.; Broniarczyk, J.; Banks, L. Papillomaviruses and Endocytic Trafficking. Int. J. Mol. Sci. 2018, 19, 2619. [Google Scholar] [CrossRef]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The Biology and Life-Cycle of Human Papillomaviruses. Vaccine 2012, 30, F55–F70. [Google Scholar] [CrossRef]
- Warren, C.J.; Xu, T.; Guo, K.; Griffin, L.M.; Westrich, J.A.; Lee, D.; Lambert, P.F.; Santiago, M.L.; Pyeon, D. APOBEC3A Functions as a Restriction Factor of Human Papillomavirus. J. Virol. 2015, 89, 688–702. [Google Scholar] [CrossRef]
- Riva, G.; Pecorari, G.; Biolatti, M.; Pautasso, S.; Lo Cigno, I.; Garzaro, M.; Dell’Oste, V.; Landolfo, S. PYHIN Genes as Potential Biomarkers for Prognosis of Human Papillomavirus-Positive or-Negative Head and Neck Squamous Cell Carcinomas. Mol. Biol. Rep. 2019, 46, 3333–3347. [Google Scholar] [CrossRef]
- Argyris, P.P.; Wilkinson, P.E.; Jarvis, M.C.; Magliocca, K.R.; Patel, M.R.; Vogel, R.I.; Gopalakrishnan, R.; Koutlas, I.G.; Harris, R.S. Endogenous APOBEC3B Overexpression Characterizes HPV-Positive and HPV-Negative Oral Epithelial Dysplasias and Head and Neck Cancers. Mod. Pathol. 2021, 34, 280–290. [Google Scholar] [CrossRef]
- Riva, G.; Albano, C.; Gugliesi, F.; Pasquero, S.; Pacheco, S.F.C.; Pecorari, G.; Landolfo, S.; Biolatti, M.; Dell’Oste, V. HPV Meets APOBEC: New Players in Head and Neck Cancer. Int. J. Mol. Sci. 2021, 22, 1402. [Google Scholar] [CrossRef]
- Mori, S.; Takeuchi, T.; Ishii, Y.; Kukimoto, I. Identification of APOBEC3B Promoter Elements Responsible for Activation by Human Papillomavirus Type 16 E6. Biochem. Biophys. Res. Commun. 2015, 460, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Takeuchi, T.; Ishii, Y.; Yugawa, T.; Kiyono, T.; Nishina, H.; Kukimoto, I. Human Papillomavirus 16 E6 Upregulates APOBEC3B via the TEAD Transcription Factor. J. Virol. 2017, 91, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Periyasamy, M.; Singh, A.K.; Gemma, C.; Kranjec, C.; Farzan, R.; Leach, D.A.; Navaratnam, N.; Pálinkás, H.L.; Vértessy, B.G.; Fenton, T.R. P53 Controls Expression of the DNA Deaminase APOBEC3B to Limit Its Potential Mutagenic Activity in Cancer Cells. Nucleic Acids Res. 2017, 45, 11056–11069. [Google Scholar] [CrossRef] [PubMed]
- Westrich, J.A.; Warren, C.J.; Klausner, M.J.; Guo, K.; Liu, C.-W.; Santiago, M.L.; Pyeon, D. Human Papillomavirus 16 E7 Stabilizes APOBEC3A Protein by Inhibiting Cullin 2-Dependent Protein Degradation. J. Virol. 2018, 92, 10–1128. [Google Scholar] [CrossRef]
- Wang, Z.; Wakae, K.; Kitamura, K.; Aoyama, S.; Liu, G.; Koura, M.; Monjurul, A.M.; Kukimoto, I.; Muramatsu, M. APOBEC3 Deaminases Induce Hypermutation in Human Papillomavirus 16 DNA upon Beta Interferon Stimulation. J. Virol. 2014, 88, 1308–1317. [Google Scholar] [CrossRef] [PubMed]
- Wakae, K.; Aoyama, S.; Wang, Z.; Kitamura, K.; Liu, G.; Monjurul, A.M.; Koura, M.; Imayasu, M.; Sakamoto, N.; Nakamura, M. Detection of Hypermutated Human Papillomavirus Type 16 Genome by Next-Generation Sequencing. Virology 2015, 485, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Faden, D.L.; Kuhs, K.A.L.; Lin, M.; Langenbucher, A.; Pinheiro, M.; Yeager, M.; Cullen, M.; Boland, J.F.; Steinberg, M.; Bass, S. APOBEC Mutagenesis Is Concordant between Tumor and Viral Genomes in HPV-Positive Head and Neck Squamous Cell Carcinoma. Viruses 2021, 13, 1666. [Google Scholar] [CrossRef] [PubMed]
- Kukimoto, I.; Mori, S.; Aoyama, S.; Wakae, K.; Muramatsu, M.; Kondo, K. Hypermutation in the E2 Gene of Human Papillomavirus Type 16 in Cervical Intraepithelial Neoplasia. J. Med. Virol. 2015, 87, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Mirabello, L.; Yeager, M.; Yu, K.; Clifford, G.M.; Xiao, Y.; Zhu, B.; Cullen, M.; Boland, J.F.; Wentzensen, N.; Nelson, C.W. HPV16 E7 Genetic Conservation Is Critical to Carcinogenesis. Cell 2017, 170, 1164–1174. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Xiao, Y.; Yeager, M.; Clifford, G.; Wentzensen, N.; Cullen, M.; Boland, J.F.; Bass, S.; Steinberg, M.K.; Raine-Bennett, T. Mutations in the HPV16 Genome Induced by APOBEC3 Are Associated with Viral Clearance. Nat. Commun. 2020, 11, 886. [Google Scholar] [CrossRef] [PubMed]
- Cook, L. Polyomaviruses. In Diagnostic Microbiology of the Immunocompromised Host; Wiley: Hoboken, NJ, USA, 2016; pp. 197–216. [Google Scholar] [CrossRef]
- Starrett, G.J.; Buck, C.B. The Case for BK Polyomavirus as a Cause of Bladder Cancer. Curr. Opin. Virol. 2019, 39, 8–15. [Google Scholar] [CrossRef]
- Verhalen, B.; Starrett, G.J.; Harris, R.S.; Jiang, M. Functional Upregulation of the DNA Cytosine Deaminase APOBEC3B by Polyomaviruses. J. Virol. 2016, 90, 6379–6386. [Google Scholar] [CrossRef]
- Starrett, G.J.; Serebrenik, A.A.; Roelofs, P.A.; McCann, J.L.; Verhalen, B.; Jarvis, M.C.; Stewart, T.A.; Law, E.K.; Krupp, A.; Jiang, M. Polyomavirus T Antigen Induces APOBEC3B Expression Using an LXCXE-Dependent and TP53-Independent Mechanism. mBio 2019, 10, 10–1128. [Google Scholar] [CrossRef]
- Peretti, A.; Geoghegan, E.M.; Pastrana, D.V.; Smola, S.; Feld, P.; Sauter, M.; Lohse, S.; Ramesh, M.; Lim, E.S.; Wang, D. Characterization of BK Polyomaviruses from Kidney Transplant Recipients Suggests a Role for APOBEC3 in Driving In-Host Virus Evolution. Cell Host Microbe 2018, 23, 628–635. [Google Scholar] [CrossRef]
- McIlroy, D.; Peltier, C.; Nguyen, M.-L.; Manceau, L.; Mobuchon, L.; Le Baut, N.; Nguyen, N.-K.; Tran, M.-C.; Nguyen, T.-C.; Bressollette-Bodin, C. Quantification of APOBEC3 Mutation Rates Affecting the VP1 Gene of BK Polyomavirus In Vivo. Viruses 2022, 14, 2077. [Google Scholar] [CrossRef]
- Liu, W.; You, J. Molecular Mechanisms of Merkel Cell Polyomavirus Transformation and Replication. Annu. Rev. Virol. 2020, 7, 289–307. [Google Scholar] [CrossRef]
- Li, J.; Wang, X.; Diaz, J.; Tsang, S.H.; Buck, C.B.; You, J. Merkel Cell Polyomavirus Large T Antigen Disrupts Host Genomic Integrity and Inhibits Cellular Proliferation. J. Virol. 2013, 87, 9173–9188. [Google Scholar] [CrossRef]
- Soikkeli, A.I.; Kyläniemi, M.K.; Sihto, H.; Alinikula, J. Oncogenic Merkel Cell Polyomavirus T Antigen Truncating Mutations Are Mediated by APOBEC3 Activity in Merkel Cell Carcinoma. Cancer Res. Commun. 2022, 2, 1344–1354. [Google Scholar] [CrossRef] [PubMed]
- Que, L.; Li, Y.; Dainichi, T.; Kukimoto, I.; Nishiyama, T.; Nakano, Y.; Shima, K.; Suzuki, T.; Sato, Y.; Horike, S. IFN-Γ–induced APOBEC3B Contributes to Merkel Cell Polyomavirus Genome Mutagenesis in Merkel Cell Carcinoma. J. Investig. Dermatol. 2022, 142, 1793–1803. [Google Scholar] [CrossRef] [PubMed]
- Cruchley, A.T.; Williams, D.M.; Niedobitek, G.; Young, L.S. Epstein-Barr Virus: Biology and Disease. Oral Dis. 1997, 3, S156–S163. [Google Scholar] [CrossRef] [PubMed]
- Kuraoka, M.; McWilliams, L.; Kelsoe, G. AID Expression during B-Cell Development: Searching for Answers. Immunol. Res. 2011, 49, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Martinez, T.; Shapiro, M.; Bhaduri-McIntosh, S.; MacCarthy, T. Evolutionary Effects of the AID/APOBEC Family of Mutagenic Enzymes on Human Gamma-Herpesviruses. Virus Evol. 2019, 5, vey040. [Google Scholar] [CrossRef]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein–Barr Virus: More than 50 Years Old and Still Providing Surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef]
- Dyson, P.J.; Farrell, P.J. Chromatin Structure of Epstein-Barr Virus. J. Gen. Virol. 1985, 66, 1931–1940. [Google Scholar] [CrossRef]
- Drake, J.W.; Hwang, C.B.C. On the Mutation Rate of Herpes Simplex Virus Type 1. Genetics 2005, 170, 969–970. [Google Scholar] [CrossRef] [PubMed]
- Tobollik, S.; Meyer, L.; Buettner, M.; Klemmer, S.; Kempkes, B.; Kremmer, E.; Niedobitek, G.; Jungnickel, B. Epstein-Barr Virus Nuclear Antigen 2 Inhibits AID Expression during EBV-Driven B-Cell Growth. Blood 2006, 108, 3859–3864. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wang, Y.; Sun, Y.; Zheng, J.; Zhu, D. MiR-155 up-Regulation by LMP1 DNA Contributes to Increased Nasopharyngeal Carcinoma Cell Proliferation and Migration. Eur. Arch. Oto-Rhino-Laryngol. 2014, 271, 1939–1945. [Google Scholar] [CrossRef]
- Bekerman, E.; Jeon, D.; Ardolino, M.; Coscoy, L. A Role for Host Activation-Induced Cytidine Deaminase in Innate Immune Defense against KSHV. PLoS Pathog. 2013, 9, e1003748. [Google Scholar] [CrossRef]
- Cheng, A.Z.; Moraes, S.N.; Attarian, C.; Yockteng-Melgar, J.; Jarvis, M.C.; Biolatti, M.; Galitska, G.; Dell’Oste, V.; Frappier, L.; Bierle, C.J. A Conserved Mechanism of APOBEC3 Relocalization by Herpesviral Ribonucleotide Reductase Large Subunits. J. Virol. 2019, 93, 10–1128. [Google Scholar] [CrossRef]
- Cheng, A.Z.; Moraes, S.N.; Shaban, N.M.; Fanunza, E.; Bierle, C.J.; Southern, P.J.; Bresnahan, W.A.; Rice, S.A.; Harris, R.S. APOBECs and Herpesviruses. Viruses 2021, 13, 390. [Google Scholar] [CrossRef] [PubMed]
- Suspene, R.; Aynaud, M.-M.; Koch, S.; Pasdeloup, D.; Labetoulle, M.; Gaertner, B.; Vartanian, J.-P.; Meyerhans, A.; Wain-Hobson, S. Genetic Editing of Herpes Simplex Virus 1 and Epstein-Barr Herpesvirus Genomes by Human APOBEC3 Cytidine Deaminases in Culture and in Vivo. J. Virol. 2011, 85, 7594–7602. [Google Scholar] [CrossRef]
- Yang, Z.; Zhuang, L.; Yu, Y.; Zhou, W.; Lu, Y.; Xu, Q.; Tang, B.; Chen, X. Overexpression of APOBEC3F in Tumor Tissues Is Potentially Predictive for Poor Recurrence-Free Survival from HBV-Related Hepatocellular Carcinoma. Discov. Med. 2015, 20, 349–356. [Google Scholar]
- Aaltonen, L.A.; Abascal, F.; Abeshouse, A.; Aburatani, H.; Adams, D.J.; Agrawal, N.; Ahn, K.S.; Ahn, S.-M.; Aikata, H.; Akbani, R.; et al. Pan-Cancer Analysis of Whole Genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef]
- Banister, C.E.; Liu, C.; Pirisi, L.; Creek, K.E.; Buckhaults, P.J. Identification and Characterization of HPV-Independent Cervical Cancers. Oncotarget 2017, 8, 13375. [Google Scholar] [CrossRef] [PubMed]
- Vieira, V.C.; Leonard, B.; White, E.A.; Starrett, G.J.; Temiz, N.A.; Lorenz, L.D.; Lee, D.; Soares, M.A.; Lambert, P.F.; Howley, P.M. Human Papillomavirus E6 Triggers Upregulation of the Antiviral and Cancer Genomic DNA Deaminase APOBEC3B. mBio 2014, 5, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.; Chakravarthy, A.; Su, X.; Boshoff, C.; Fenton, T.R. APOBEC-Mediated Cytosine Deamination Links PIK3CA Helical Domain Mutations to Human Papillomavirus-Driven Tumor Development. Cell Rep. 2014, 7, 1833–1841. [Google Scholar] [CrossRef] [PubMed]
- Knepper, T.C.; Montesion, M.; Russell, J.S.; Sokol, E.S.; Frampton, G.M.; Miller, V.A.; Albacker, L.A.; McLeod, H.L.; Eroglu, Z.; Khushalani, N.I. The Genomic Landscape of Merkel Cell Carcinoma and Clinicogenomic Biomarkers of Response to Immune Checkpoint Inhibitor Therapy. Clin. Cancer Res. 2019, 25, 5961–5971. [Google Scholar] [CrossRef] [PubMed]
- Rao, N.; Starrett, G.J.; Piaskowski, M.L.; Butler, K.E.; Golubeva, Y.; Yan, W.; Lawrence, S.M.; Dean, M.; Garcia-Closas, M.; Baris, D. Analysis of Several Common APOBEC-Type Mutations in Bladder Tumors Suggests Links to Viral Infection. Cancer Prev. Res. 2023, 16, 561–570. [Google Scholar] [CrossRef]
- Leonard, B.; Hart, S.N.; Burns, M.B.; Carpenter, M.A.; Temiz, N.A.; Rathore, A.; Vogel, R.I.; Nikas, J.B.; Law, E.K.; Brown, W.L. APOBEC3B Upregulation and Genomic Mutation Patterns in Serous Ovarian Carcinoma. Cancer Res. 2013, 73, 7222–7231. [Google Scholar] [CrossRef]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.V.; Carter, S.L.; Saksena, G. An APOBEC Cytidine Deaminase Mutagenesis Pattern Is Widespread in Human Cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; NiccoloBolli, A.B.; Børresen-Dale, A.-L.; Boyault, S. Corrigendum: Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar]
- Petljak, M.; Dananberg, A.; Chu, K.; Bergstrom, E.N.; Striepen, J.; von Morgen, P.; Chen, Y.; Shah, H.; Sale, J.E.; Alexandrov, L.B. Mechanisms of APOBEC3 Mutagenesis in Human Cancer Cells. Nature 2022, 607, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Roberts, S.A.; Klimczak, L.J.; Sterling, J.F.; Saini, N.; Malc, E.P.; Kim, J.; Kwiatkowski, D.J.; Fargo, D.C.; Mieczkowski, P.A. An APOBEC3A Hypermutation Signature Is Distinguishable from the Signature of Background Mutagenesis by APOBEC3B in Human Cancers. Nat. Genet. 2015, 47, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Langenbucher, A.; Bowen, D.; Sakhtemani, R.; Bournique, E.; Wise, J.F.; Zou, L.; Bhagwat, A.S.; Buisson, R.; Lawrence, M.S. An Extended APOBEC3A Mutation Signature in Cancer. Nat. Commun. 2021, 12, 1602. [Google Scholar] [CrossRef]
- Burns, M.B.; Temiz, N.A.; Harris, R.S. Evidence for APOBEC3B Mutagenesis in Multiple Human Cancers. Nat. Genet. 2013, 45, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, M.A.; Temiz, N.A.; Ibrahim, M.A.; Jarvis, M.C.; Brown, M.R.; Argyris, P.P.; Brown, W.L.; Starrett, G.J.; Yee, D.; Harris, R.S. Mutational Impact of APOBEC3A and APOBEC3B in a Human Cell Line and Comparisons to Breast Cancer. PLoS Genet. 2023, 19, e1011043. [Google Scholar] [CrossRef] [PubMed]
- Network, C.G.A.R. Integrated Genomic and Molecular Characterization of Cervical Cancer. Nature 2017, 543, 378. [Google Scholar]
- Ojesina, A.I.; Lichtenstein, L.; Freeman, S.S.; Pedamallu, C.S.; Imaz-Rosshandler, I.; Pugh, T.J.; Cherniack, A.D.; Ambrogio, L.; Cibulskis, K.; Bertelsen, B. Landscape of Genomic Alterations in Cervical Carcinomas. Nature 2014, 506, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Lou, H.; Villagran, G.; Boland, J.F.; Im, K.M.; Polo, S.; Zhou, W.; Odey, U.; Juárez-Torres, E.; Medina-Martínez, I.; Roman-Basaure, E. Genome Analysis of Latin American Cervical Cancer: Frequent Activation of the PIK3CA Pathway. Clin. Cancer Res. 2015, 21, 5360–5370. [Google Scholar] [CrossRef] [PubMed]
- Koncar, R.F.; Feldman, R.; Bahassi, E.M.; Hashemi Sadraei, N. Comparative Molecular Profiling of HPV-induced Squamous Cell Carcinomas. Cancer Med. 2017, 6, 1673–1685. [Google Scholar] [CrossRef]
- Faden, D.L.; Ding, F.; Lin, Y.; Zhai, S.; Kuo, F.; Chan, T.A.; Morris, L.G.; Ferris, R.L. APOBEC Mutagenesis Is Tightly Linked to the Immune Landscape and Immunotherapy Biomarkers in Head and Neck Squamous Cell Carcinoma. Oral Oncol. 2019, 96, 140–147. [Google Scholar] [CrossRef]
- Wolf, Y.; Bartok, O.; Patkar, S.; Eli, G.B.; Cohen, S.; Litchfield, K.; Levy, R.; Jiménez-Sánchez, A.; Trabish, S.; Lee, J.S. UVB-Induced Tumor Heterogeneity Diminishes Immune Response in Melanoma. Cell 2019, 179, 219–235. [Google Scholar] [CrossRef]
- Eberhardt, C.S.; Kissick, H.T.; Patel, M.R.; Cardenas, M.A.; Prokhnevska, N.; Obeng, R.C.; Nasti, T.H.; Griffith, C.C.; Im, S.J.; Wang, X. Functional HPV-Specific PD-1+ Stem-like CD8 T Cells in Head and Neck Cancer. Nature 2021, 597, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Bobrovnitchaia, I.; Valieris, R.; Drummond, R.D.; Lima, J.P.; Freitas, H.C.; Bartelli, T.F.; de Amorim, M.G.; Nunes, D.N.; Dias-Neto, E.; da Silva, I.T. APOBEC-mediated DNA Alterations: A Possible New Mechanism of Carcinogenesis in EBV-positive Gastric Cancer. Int. J. Cancer 2020, 146, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Starrett, G.J.; Marcelus, C.; Cantalupo, P.G.; Katz, J.P.; Cheng, J.; Akagi, K.; Thakuria, M.; Rabinowits, G.; Wang, L.C.; Symer, D.E. Merkel Cell Polyomavirus Exhibits Dominant Control of the Tumor Genome and Transcriptome in Virus-Associated Merkel Cell Carcinoma. mBio 2017, 8, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F. Exome Sequencing of Hepatocellular Carcinomas Identifies New Mutational Signatures and Potential Therapeutic Targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Lu, Y.; Xu, Q.; Zhuang, L.; Tang, B.; Chen, X. Correlation of APOBEC3 in Tumor Tissues with Clinico-Pathological Features and Survival from Hepatocellular Carcinoma after Curative Hepatectomy. Int. J. Clin. Exp. Med. 2015, 8, 7762. [Google Scholar] [PubMed]
- Xu, R.; Zhang, X.; Zhang, W.; Fang, Y.; Zheng, S.; Yu, X. Association of Human APOBEC3 Cytidine Deaminases with the Generation of Hepatitis Virus B x Antigen Mutants and Hepatocellular Carcinoma. Hepatology 2007, 46, 1810–1820. [Google Scholar] [CrossRef]
- Ma, W.; Ho, D.W.; Sze, K.M.; Tsui, Y.; Chan, L.; Lee, J.M.; Ng, I.O. APOBEC3B Promotes Hepatocarcinogenesis and Metastasis through Novel Deaminase-independent Activity. Mol. Carcinog. 2019, 58, 643–653. [Google Scholar] [CrossRef]
- Li, S.; Bao, X.; Wang, D.; You, L.; Li, X.; Yang, H.; Bian, J.; Wang, Y.; Yang, Y. APOBEC3B and IL-6 Form a Positive Feedback Loop in Hepatocellular Carcinoma Cells. Sci. China Life Sci. 2017, 60, 617–626. [Google Scholar] [CrossRef]
- Wang, D.; Li, X.; Li, J.; Lu, Y.; Zhao, S.; Tang, X.; Chen, X.; Li, J.; Zheng, Y.; Li, S. APOBEC3B Interaction with PRC2 Modulates Microenvironment to Promote HCC Progression. Gut 2019, 68, gutjnl-2018. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| DNA Tumour Virus | Viral Genome Editing by A3/AID a | Restriction of Infection | A3 Expression in Tumours | A3 Mutational Signatures in Cancer | Mutations in Cancer Driver Genes/Viral Oncogenes | References | |
|---|---|---|---|---|---|---|---|
| Hepadnaviruses | HBV | Localised: A3A, A3B, A3C, A3D, A3F, A3G, A3H | A3B, A3C, A3F, A3G, A3H/hapII | A3B, A3D, A3F, A3G, A3H | Low | Unknown/HBx | [6,59,62,63,64,70,72,73,74,76,77,78,119,120] |
| Papillomaviruses | HPV | Genome-wide: A3A, A3B, A3H | A3A | A3A, A3B | Strong: A3A, A3B | PIK3CA, FBXW7, EP300, MAPK1, TP53, ERBB2, NFE2L2, KRAS,… | [41,84,85,86,92,93,95,121,122,123,124] |
| Polyomaviruses | MCPyV | Genome-wide: A3A, A3B, A3G | Unknown | A3A, A3B and A3H | Low | Unknown/LT antigen | [99,106,107,125] |
| BKPyV | Genome-wide: A3A, A3B | Unknown | Unknown | Unknown | GFR3/PIK3CA b | [100,102,103,126] | |
| Gamma-herpesviruses | EBV | Localised: A3B (A3H), AID | Unknown (A3C in HSV-1) | A3B, A3C, A3D, A3F, A3G, A3H | EBV-positive breast and gastric cancers | PIK3CA and some other oncogenes | [7,8,110,119] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lovšin, N.; Gangupam, B.; Bergant Marušič, M. The Intricate Interplay between APOBEC3 Proteins and DNA Tumour Viruses. Pathogens 2024, 13, 187. https://doi.org/10.3390/pathogens13030187
Lovšin N, Gangupam B, Bergant Marušič M. The Intricate Interplay between APOBEC3 Proteins and DNA Tumour Viruses. Pathogens. 2024; 13(3):187. https://doi.org/10.3390/pathogens13030187
Chicago/Turabian StyleLovšin, Nika, Bhavani Gangupam, and Martina Bergant Marušič. 2024. "The Intricate Interplay between APOBEC3 Proteins and DNA Tumour Viruses" Pathogens 13, no. 3: 187. https://doi.org/10.3390/pathogens13030187
APA StyleLovšin, N., Gangupam, B., & Bergant Marušič, M. (2024). The Intricate Interplay between APOBEC3 Proteins and DNA Tumour Viruses. Pathogens, 13(3), 187. https://doi.org/10.3390/pathogens13030187