
Chromosome-Level Assemblies for the Pine Pitch Canker Pathogen Fusarium circinatum

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Genome Sequencing and Assembly
2.2. Evaluation of Genome Quality and Completeness
2.3. Genome Annotation
2.4. Compilation of Ecological and Biological Metadata for FSP34 and KS17
3. Results
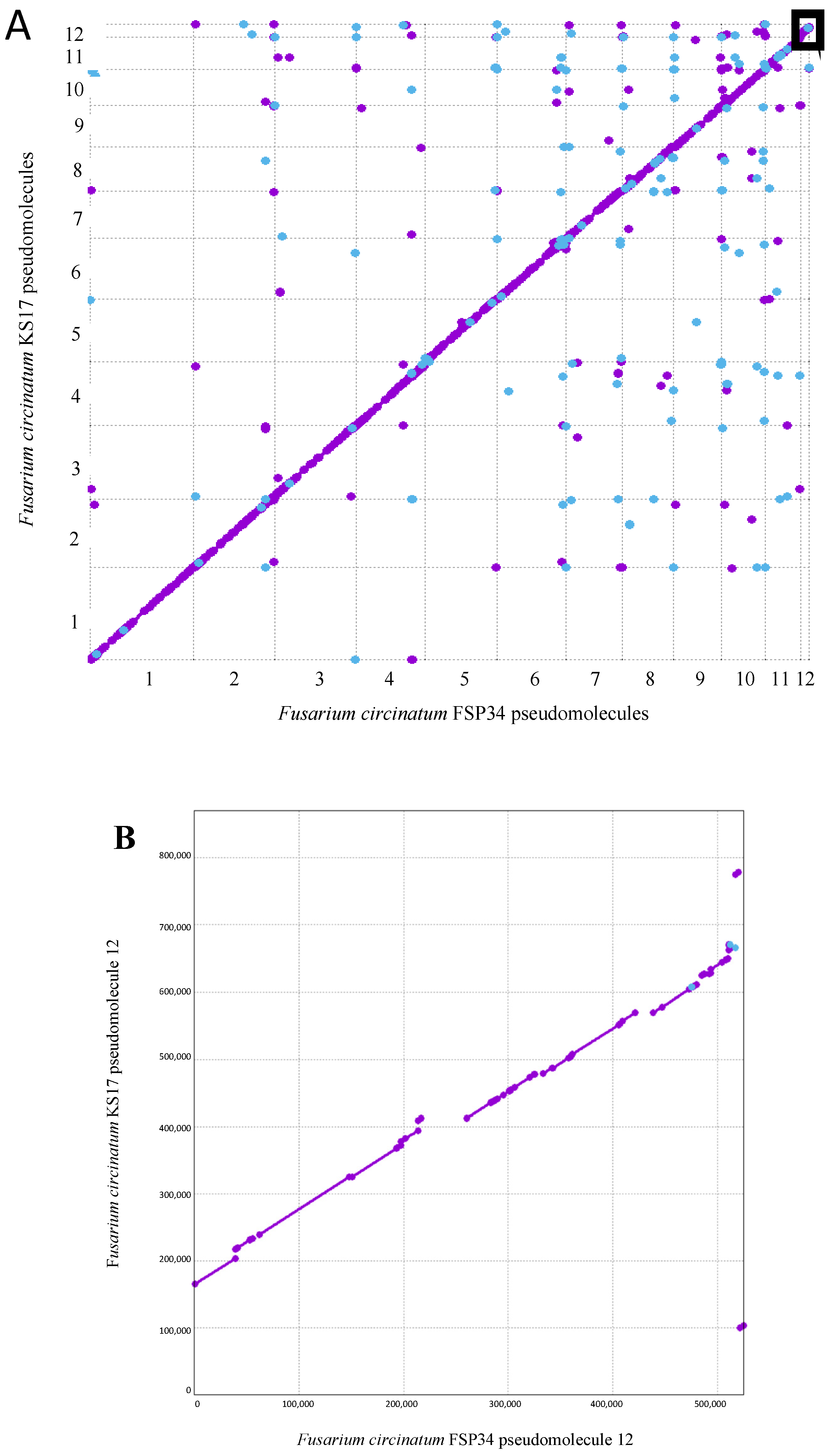
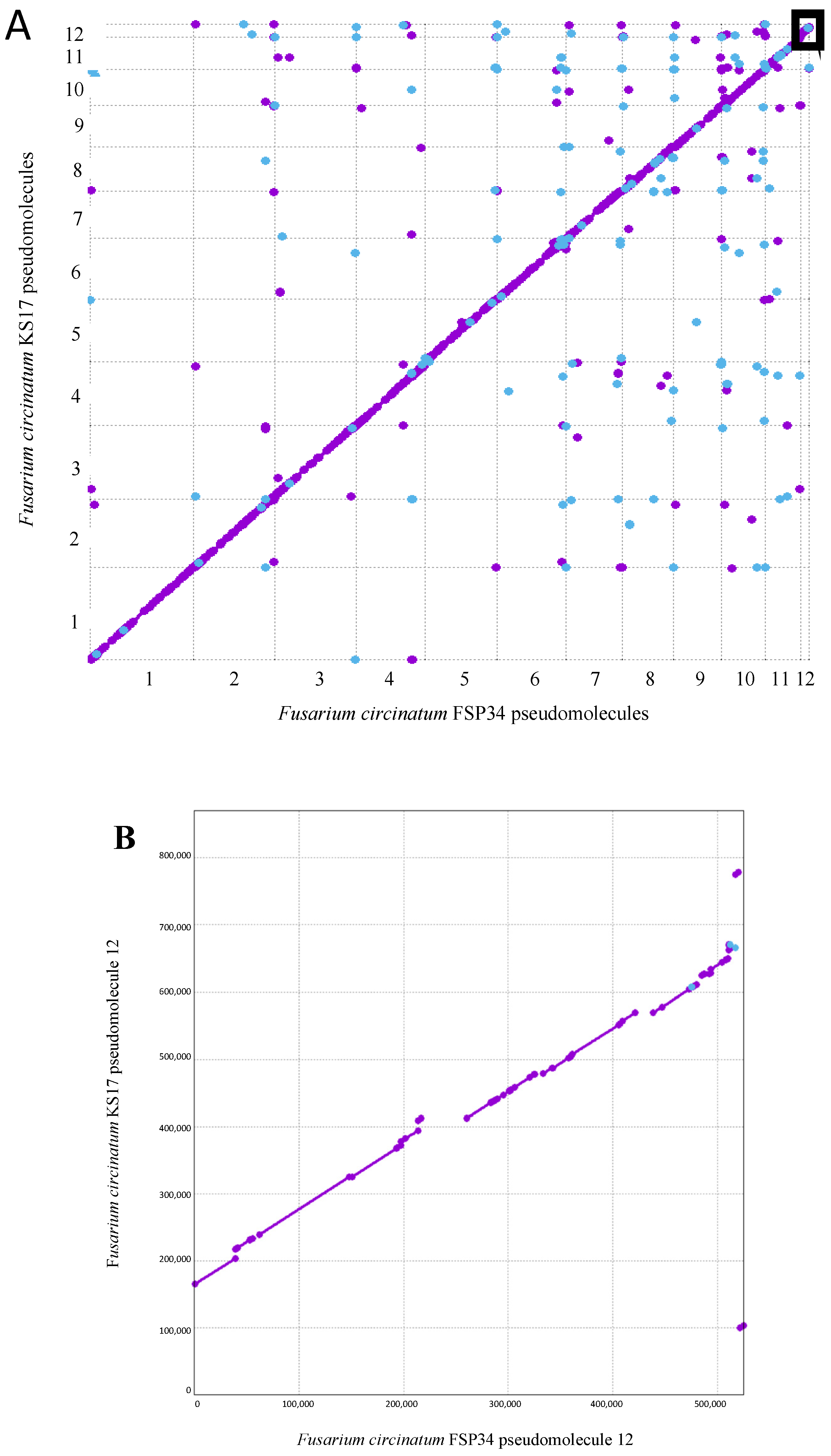
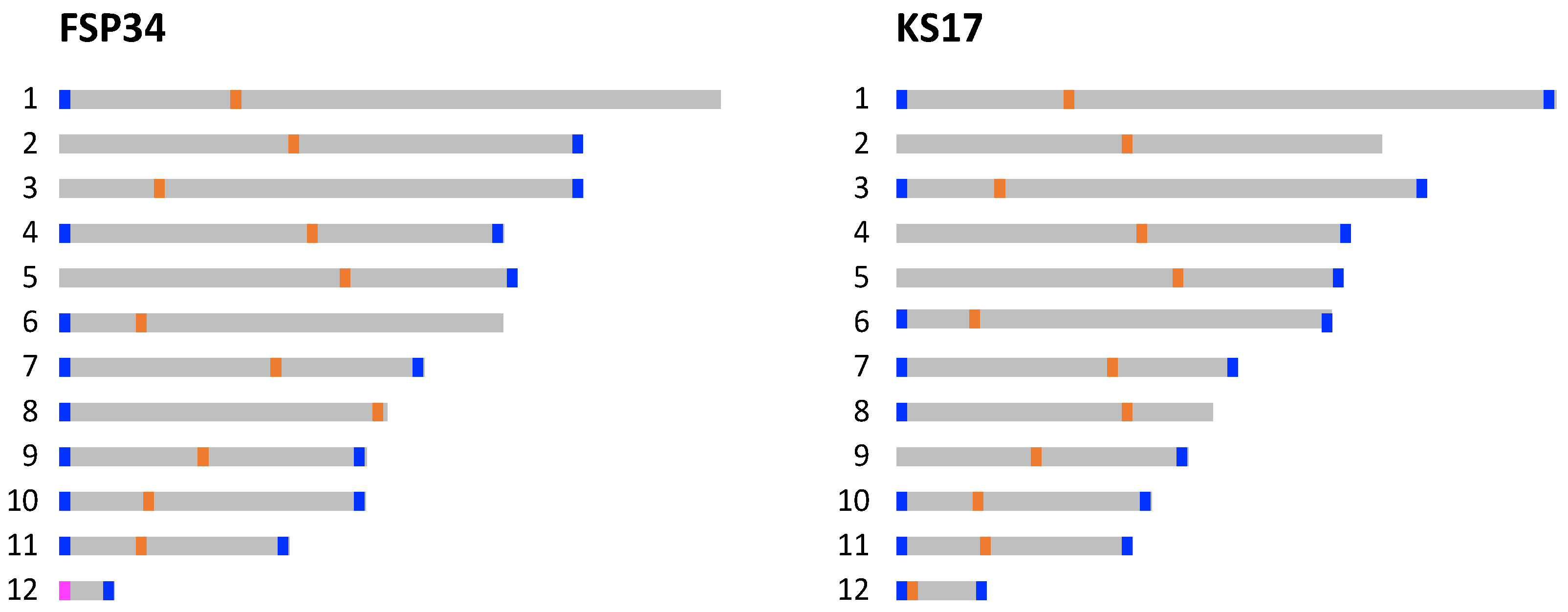
3.1. Chromosome-Level Assemblies for FSP34 and KS17
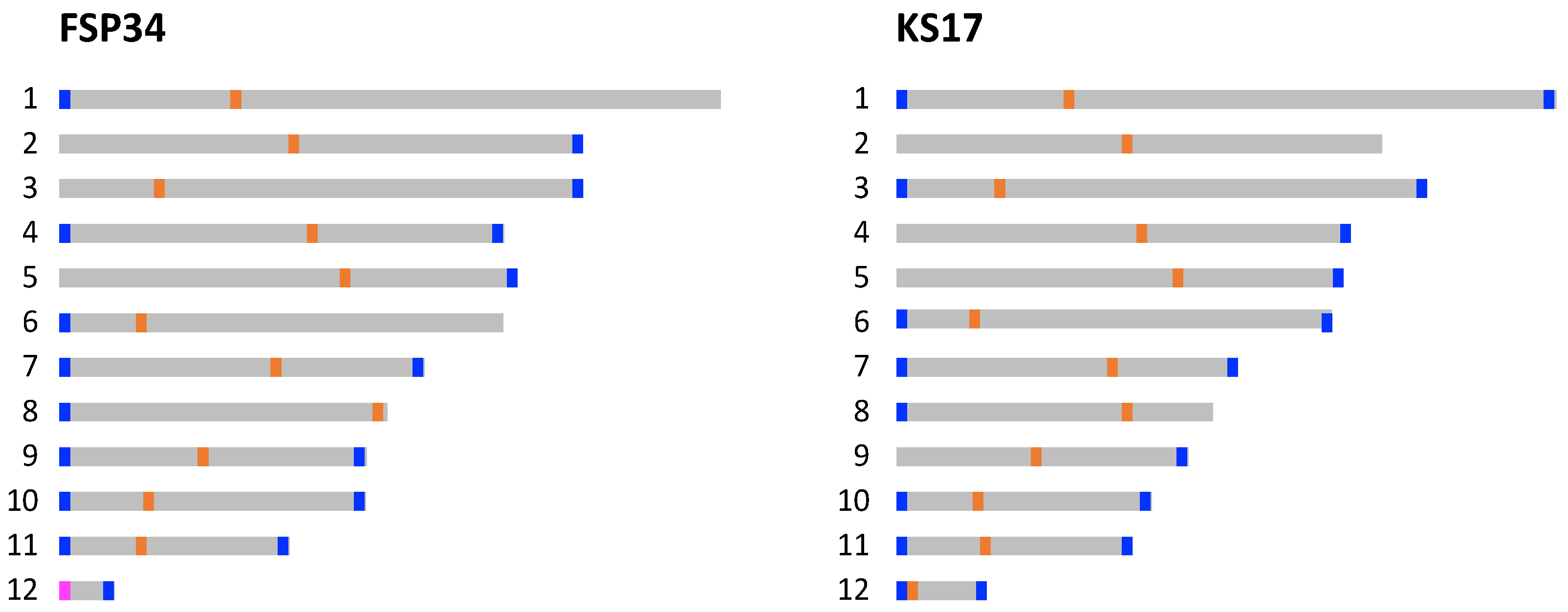
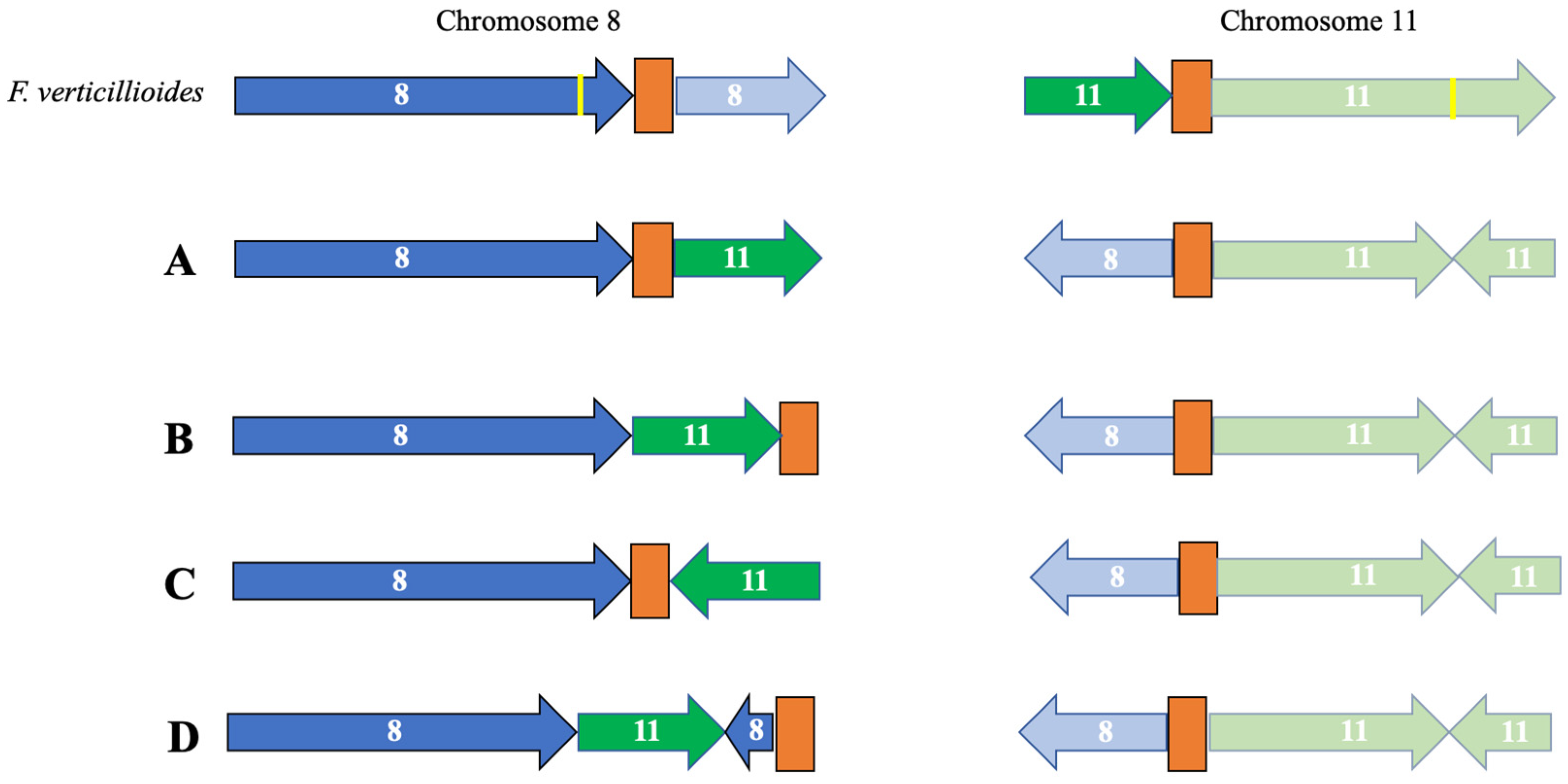
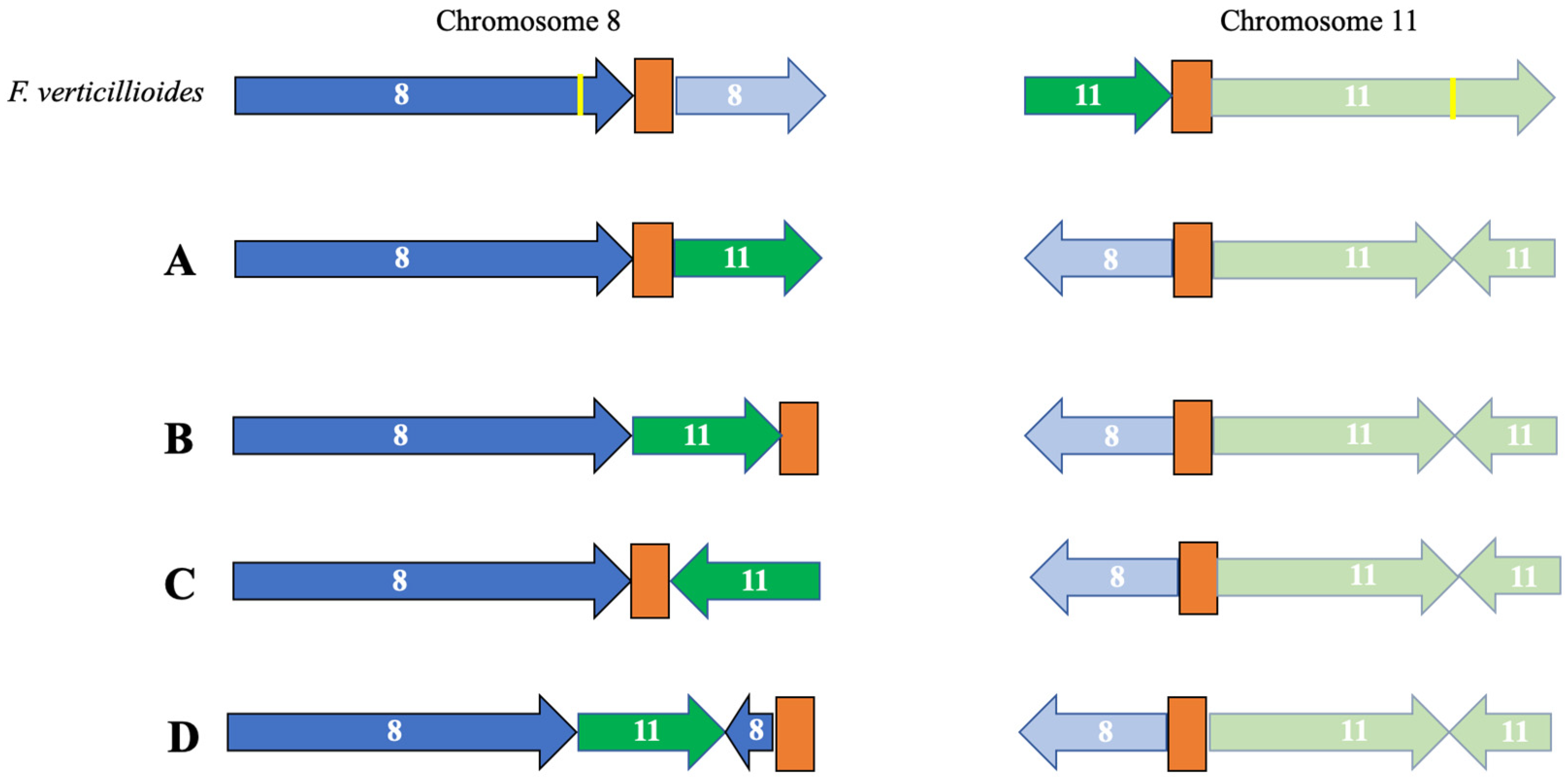
3.2. Identification of Telomeres and Centromeres
3.3. Genome Completeness and Gene Content
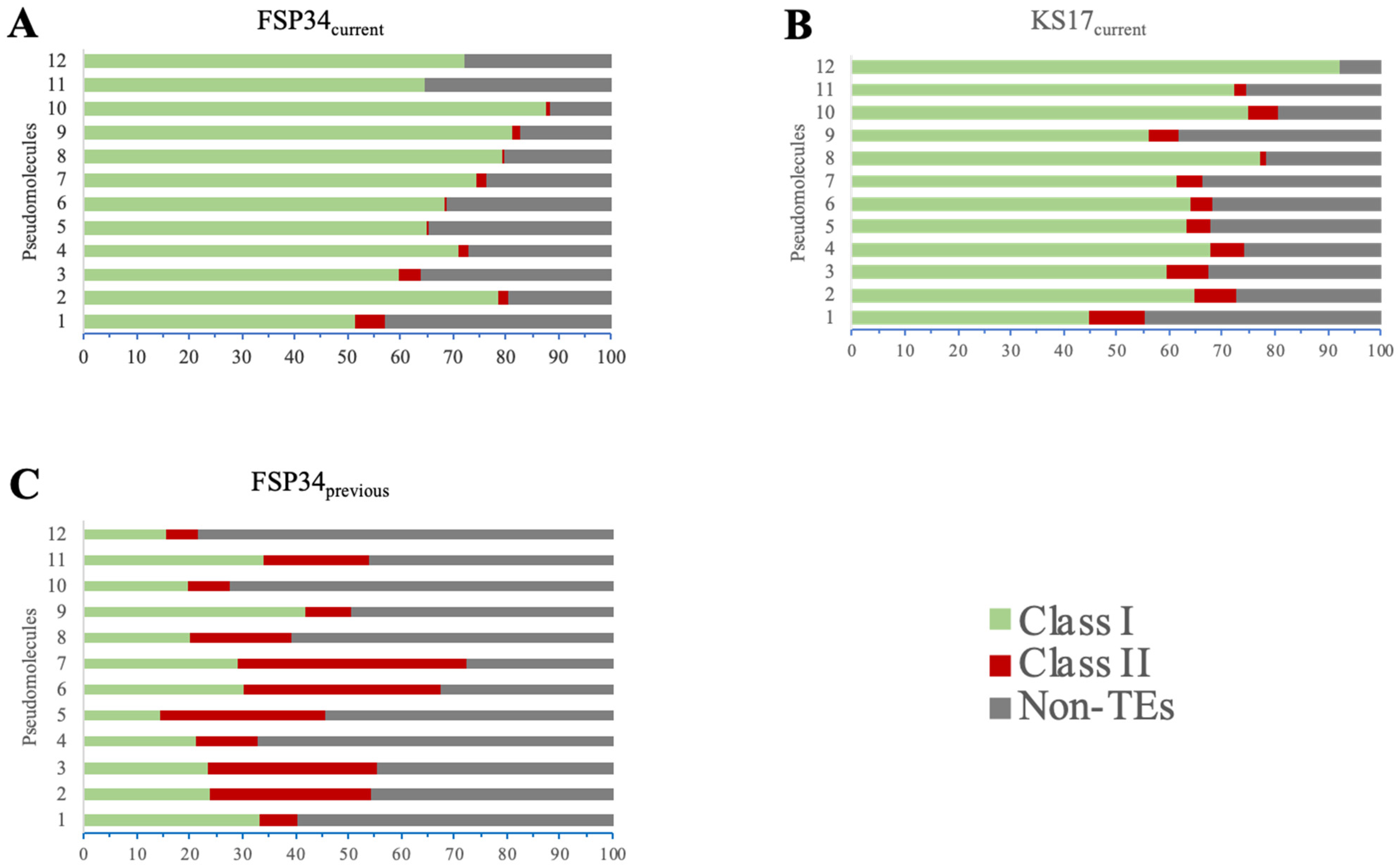
3.4. Genome Repetitiveness and TE Content
3.5. Detailed Source and Biological Information for KS17 and FSP34
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Engel, S.R.; Dietrich, F.S.; Fisk, D.G.; Binkley, G.; Balakrishnan, R.; Costanzo, M.C.; Dwight, S.S.; Hitz, B.C.; Karra, K.; Nash, R.S.; et al. The reference genome sequence of Saccharomyces cerevisiae: Then and Now. G3 Genes Genomes Genet. 2014, 4, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Galagan, J.E.; Calvo, S.E.; Borkovich, K.A.; Selker, E.U.; Read, N.D.; Jaffe, D.; FitzHugh, W.; Ma, L.-J.; Smirnov, S.; Purcell, S.; et al. The genome sequence of the filamentous fungus Neurospora crassa. Nature 2003, 422, 859–868. [Google Scholar] [CrossRef]
- Grigoriev, I.V.; Nikitin, R.; Haridas, S.; Kuo, A.; Ohm, R.; Otillar, R.; Riley, R.; Salamov, A.; Zhao, X.; Korzeniewski, F.; et al. MycoCosm portal: Gearing up for 1000 fungal genomes. Nucleic Acids Res. 2014, 42, D699–D704. [Google Scholar] [CrossRef]
- Ma, L.-J.; Van der Does, H.C.; Borkovich, K.A.; Coleman, J.J.; Daboussi, M.-J.; Di Pietro, A.; Dufresne, M.; Freitag, M.; Grabherr, M.; Henrissat, B.; et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 2010, 464, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.K. Fungal genome sequencing: Basic biology to biotechnology. Crit. Rev. Biotechnol. 2016, 36, 743–759. [Google Scholar] [CrossRef]
- Miyauchi, S.; Kiss, E.; Kuo, A.; Drula, E.; Kohler, A.; Sánchez-García, M.; Morin, E.; Andreopoulos, B.; Barry, K.W.; Bonito, G.; et al. Large-scale genome sequencing of mycorrhizal fungi provides insights into the early evolution of symbiotic traits. Nat. Commun. 2021, 11, 5125. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.-J.; Geiser, D.M.; Proctor, R.; Rooney, A.P.; O’Donnell, K.; Trail, F.; Gardiner, D.M.; Manners, J.M.; Kazan, K. Fusarium pathogenomics. Annu. Rev. Microbiol. 2013, 67, 399–416. [Google Scholar] [CrossRef]
- Delulio, G.A.; Guo, L.; Zhang, Y.; Goldberg, J.M. Kinome expansion in the Fusarium oxysporum species complex driven by accessory chromosomes. mSphere 2018, 3, e00231-00218. [Google Scholar]
- Li, Y.; Steenwyk, J.L.; Chang, Y.; Wang, Y.; James, T.Y.; Stajich, J.E.; Spatafora, J.W.; Groenewald, M.; Dunn, C.W.; Hittinger, C.T.; et al. A genome-scale phylogeny of the kingdom Fungi. Curr. Biol. 2021, 31, 1663–1665.e5. [Google Scholar] [CrossRef]
- Wiemann, P.; Sieber, C.M.K.; Von Bargen, K.W.; Studt, L.; Niehaus, E.-M.; Hub, K.; Michielse, C.B.; Albermann, S.; Wagner, D.; Espino, J.J.; et al. Unleashing the cryptic genome: Genome-wide analyses of the rice pathogen Fusarium fujikuroi reveal complex regulation of secondary metabolism and novel metabolites. PLoS Pathog. 2013, 9, e1003475. [Google Scholar] [CrossRef]
- Fokkens, L.; Guo, L.; Dora, S.; Wang, B.; Ye, K.; Sánchez-Rodríguez, C.; Croll, D. A chromosome-scale genome assembly for the Fusarium oxysporum strain Fo5176 to establish a model Arabidopsis-fungal pathosystem. G3 Genes Genomes Genet. 2020, 10, 3549–3555. [Google Scholar] [CrossRef] [PubMed]
- Niehaus, E.-M.; Münsterkötter, M.; Proctor, R.H.; Brown, D.W.; Sharon, A.; Idan, Y.; Oren-Young, L.; Sieber, C.M.; Novák, O.; Pĕnčík, A.; et al. Comparative “omics”of the Fusarium fujikuroi species complex highlights differences in genetic potential and metabolite synthesis. Genome Biol. Evol. 2017, 8, 3574–3599. [Google Scholar] [CrossRef] [PubMed]
- Geiser, D.M.; Aoki, T.; Bacon, C.W.; Baker, S.E.; Bhattacharyya, M.K.K.; Brandt, M.E.; Brown, D.W.; Burgess, L.W.; Chulze, S.N.; Coleman, J.J.; et al. One fungus, one name: Defining the genus Fusarium in a scientifically robust way that preserves longstanding use. Phytopathology 2013, 103, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Drenkhan, R.; Ganley, B.; Martín-García, J.; Vahalík, P.; Adamson, K.; Adamčíková, K.; Ahumada, R.; Blank, L.; Bragança, H.; Capretti, P.; et al. Global geographic distribution and host range of Fusarium circinatum, the causal agent of pine pitch canker. Forests 2020, 11, 724. [Google Scholar] [CrossRef]
- Wingfield, B.D.; Steenkamp, E.T.; Santana, Q.C.; Coetzee, M.P.A.; Bam, S.; Barnes, I.; Beukes, C.W.; Chan, W.Y.; De Vos, L.; Fourie, G.; et al. First fungal genome sequence from Africa: A preliminary analysis. S. Afr. J. Sci. 2012, 108, 1–9. [Google Scholar] [CrossRef]
- Wingfield, B.D.; Liu, M.; Nguyen, H.D.T.; Lane, F.A.; Morgan, S.W.; De Vos, L.; Wilken, P.M.; Duong, T.A.; Aylward, J.; Coetzee, M.P.A.; et al. Nine draft genome sequences of Claviceps purpurea s. lat., including C. arundinis, C. humidiphila, and C. cf. spartinea, pseudomolecules for the pitch canker pathogen Fusarium circinatum, draft genome of Davidsoniella eucalypti, Grosmannia galeiformis, Quambalaria euclaypti, and Teratospahaeria destructans. IMA Fungus 2018, 9, 401–418. [Google Scholar]
- van Wyk, S.; Wingfield, B.D.; De Vos, L.; Santana, Q.C.; Van der Merwe, N.A.; Steenkamp, E.T. Multiple independent origins for a subtelomeric locus associated with growth rate in Fusarium circinatum. IMA Fungus 2018, 9, 27–36. [Google Scholar] [CrossRef]
- De Vos, L.; Steenkamp, E.T.; Martin, S.H.; Santana, Q.C.; Fourie, G.; Van der Merwe, N.A.; Wingfield, M.J.; Wingfield, B.D. Genome-wide macrosynteny among Fusarium species within the Gibberella fujikuroi complex revealed by amplified fragment length polymorphisms. PLoS ONE 2014, 9, e114682. [Google Scholar] [CrossRef]
- Clarke, J.; Wu, H.-C.; Jayasinghe, L.; Patel, A.; Reid, S.; Bayley, H. Continuous base identification for single-molecule nanopore DNA sequencing. Nat. Nanotechnol. 2009, 4, 265–270. [Google Scholar] [CrossRef]
- Payne, A.; Holmes, N.; Rakyan, V.; Loose, M. BulkVis: A graphical viewer for Oxford nanopore bulk FAST5 files. Bioinformatics 2019, 35, 2193–2198. [Google Scholar] [CrossRef]
- Laver, T.; Harrison, J.; O’Neill, P.A.; Moore, K.; Farbos, A.; Paszkiewicz, K.; Studholme, D.J. Assessing the performance of the Oxford Nanopore Technologies MinION. Biomol. Detect. Quantif. 2015, 3, 1–8. [Google Scholar] [CrossRef]
- Saud, Z.; Kortsinoglou, A.M.; Kouvelis, V.N.; Butt, T.M. Telomere length de novo assembly of all 7 chromosomes and mitogenome sequencing of the model entomopathogenic fungus, Metarhizium brunneum, by means of a novel assembly pipeline. BMC Genom. 2021, 22, 87. [Google Scholar] [CrossRef]
- Wang, B.; Liang, X.; Gleason, M.L.; Hsiang, T.; Zhang, R.; Sun, G. A chromosome-scale assembly of the smallest Dothideomycete genome reveals a unique genome compaction mechanism in filamentous fungi. BMC Genom. 2020, 21, 321. [Google Scholar] [CrossRef]
- Crestana, G.S.; Taniguti, L.M.; dos Santos, C.P.; Benevenuto, J.; Ceresini, P.C.; Carvalho, G.; Kitajima, J.P.; Monteiro-Vitorello, C.B. Complete chromosome-scale genome sequence resource for Sporisorium panici-leucophaei, the causal agent of sourgrass smut disease. Mol. Plant-Microbe Interact. 2021, 34, 448–452. [Google Scholar] [CrossRef]
- Wang, B.; Yu, H.; Jia, Y.; Dong, Q.; Steinberg, C.; Alabouvette, C.; Edel-Hermann, V.; Kistler, H.C.; Ye, K.; Ma, L.-J.; et al. Chromosome-scale genome assembly of Fusarium oxysporum strain Fo47, a fungal endophyte and biocontrol agent. Mol. Plant-Microbe Interact. 2020, 33, 1108–1111. [Google Scholar] [CrossRef]
- McKenzie, S.K.; Walston, R.F.; Allen, J.L. Complete, high-quality genomes from long-read metagenomic sequencing of two wolf lichen thalli reveals enigmatic genome architecture. Genomics 2020, 112, 3150–3156. [Google Scholar] [CrossRef]
- Maphosa, M.N.; Steenkamp, E.T.; Kanzi, A.M.; van Wyk, S.; De Vos, L.; Santana, Q.C.; Duong, T.A.; Wingfield, B.D. Intra-species genomic variation in the pine pathogen Fusarium circinatum. J. Fungi 2022, 8, 657. [Google Scholar] [CrossRef]
- Lofgren, L.A.; Stajich, J.E. Fungal biodiversity and conservation mycology in light of new technology, big data, and changing attitudes. Curr. Biol. 2021, 31, R1312–R1325. [Google Scholar] [CrossRef]
- Sheffield, N.C.; LeRoy, N.J.; Khoroshevskyi, O. Challenges to sharing sample metadata in computational genomics. Front. Genet. 2023, 14, 154198. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillipy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Harris, R.S. Improved Pairwise Alignment of Genomic DNA; Pennsylvania State University: State College, PA, USA, 2007. [Google Scholar]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinform. Appl. Note 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Subgroup, G.P.D.P. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef]
- O’Donnell, K.; Cigelnik, E.; Nirenberg, H.I. Molecular systematics and phylogeography of the Gibberella fujikuroi species complex. Mycologia 1998, 90, 465–493. [Google Scholar] [CrossRef]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2017, 35, 543–548. [Google Scholar] [CrossRef]
- Wu, C.; Kim, Y.-S.; Smith, K.M.; Li, W.; Hood, H.M.; Staben, C.; Selker, E.U.; Sachs, M.S.; Farman, M.L. Characterization of chromosome ends in the filamentous fungus Neurospora crassa. Genetics 2009, 181, 1129–1145. [Google Scholar] [CrossRef]
- van Wyk, S.; Harrison, C.H.; Wingfield, B.D.; De Vos, L.; van der Merwe, N.A.; Steenkamp, E.T. The RIPper, a web-based tool for genome-wide quantification of Repeat-Induced Point (RIP) mutations. PeerJ 2019, 7, e7447. [Google Scholar] [CrossRef]
- Levan, A.; Fredga, K.; Sandberg, A.A. Nomenclature for centromeric position on chromosomes. Hereditas 1964, 201–220. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Korf, I.; Robb, S.M.C.; Parra, G.; Ross, E.; Moore, B.; Holt, C.; Alvarado, A.S.; Yandell, M. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008, 18, 188–196. [Google Scholar] [CrossRef]
- Stanke, M.; Schöffmann, O.; Morgenstern, B.; Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinform. 2006, 7, 62. [Google Scholar] [CrossRef]
- Ter-Hovhannisyan, V.; Lomsadze, A.; Chernoff, Y.O.; Borodovsky, M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008, 18, 1979–1990. [Google Scholar] [CrossRef]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]
- Flutre, T.; Dupart, E.; Feuillet, C.; Quesneville, H. Considering transposable element diversification in de novo annotation approaches. PLoS ONE 2011, 6, e16526. [Google Scholar] [CrossRef]
- Quesneville, H.; Bergman, C.M.; Andrieu, O.; Autard, D.; Nouaud, D.; Ashburner, M.; Anxolabehere, D. Combined evidence annotation of transposable elements in genome sequences. PLoS Comput. Biol. 2005, 1, e22. [Google Scholar] [CrossRef]
- Steenkamp, E.T.; Makhari, O.M.; Coutinho, T.A.; Wingfield, B.D.; Wingfield, M.J. Evidence for a new introduction of the pitch canker fungus Fusarium circinatum in South Africa. Plant Pathol. 2014, 63, 530–538. [Google Scholar] [CrossRef]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Gordon, T.R.; Storer, A.J.; Okamoto, D. Population structure of the pitch canker pathogen, Fusarium subglutinans f. sp. pini, in California. Mycol. Res. 1996, 100, 850–854. [Google Scholar] [CrossRef]
- Desjardins, A.E.; Plattner, R.D.; Gordon, T.R. Gibberella fujikuroi mating population A and Fusarium subglutinans from teosinte species and maize from Mexico and Central America. Mycol. Res. 2000, 104, 865–872. [Google Scholar] [CrossRef]
- McCain, A.H.; Koehler, C.S.; Tjosvold, S.A. Pitch canker threatens California pines. Calif. Agric. 1987, 41, 22–23. [Google Scholar]
- Van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The third revolution in sequencing technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef]
- Kumar, K.R.; Cowley, M.J.; Davis, R.L. Next-generation sequencing and emerging technologies. Semin. Thromb. Homestasis 2019, 45, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Coghlan, A.; Eichler, E.E.; Oliver, S.G.; Paterson, A.H.; Stein, L. Chromosome evolution in eukaryotes: A multi-kingdom perspective. Trends Genet. 2005, 21, 673–682. [Google Scholar] [CrossRef]
- Guin, K.; Sreekumar, L.; Sanyal, K. Implications of the evolutionary trajectory of centromeres in the fungal kingdom. Annu. Rev. Microbiol. 2020, 74, 835–853. [Google Scholar] [CrossRef]
- Sun, S.; Yadav, V.; Billmyre, R.B.; Cuomo, C.A.; Nowrousian, M.; Wang, L.; Souciet, J.-L.; Boekhout, T.; Porcel, B.; Wincker, P.; et al. Fungal genome and mating system transitions facilitated by chromosomal translocations involving intercentromeric recombination. PLoS Biol. 2017, 15, e2002527. [Google Scholar] [CrossRef]
- Yadav, V.; Sun, S.; Coelho, M.A.; Heitmans, J. Centromere scission drives chromosome shuffling and reproductive isolation. Proc. Natl. Acad. Sci. USA 2020, 117, 7917–7928. [Google Scholar] [CrossRef]
- Rao, S.; Sharda, S.; Oddi, V.; Nandineni, M.R. The landscape of repetitive elements in the refined genome of chilli anthracnose fungus Collectotrichum truncatum. Front. Microbiol. 2018, 9, 2367. [Google Scholar] [CrossRef]
- Stukenbrock, E.H.; Croll, D. The evolving fungal genome. Fungal Biol. Rev. 2014, 28, 1–12. [Google Scholar] [CrossRef]
- Depotter, J.R.L.; Ökmen, B.; Ebert, M.K.; Beckers, J.; Kruse, J.; Thines, M.; Doehlemann, G. High nucleotide substitution rates associated with retrotransposon proliferation. Microbiol. Spectr. 2022, 10, e0034922. [Google Scholar] [CrossRef] [PubMed]
- Malewski, T.; Matić, S.; Okorski, A.; Borowik, P.; Oszako, T. Annotation of the 12th chromosome of the forest pathogen Fusarium circinatum. Agronomy 2023, 13, 773. [Google Scholar] [CrossRef]
- Waalwijk, C.; Taga, M.; Zheng, S.-L.; Proctor, R.H.; Vaughan, M.M.; O’Donnell, K. Karyotype evolution in Fusarium. IMA Fungus 2018, 9, 13–26. [Google Scholar] [CrossRef]
- Xu, J.-R.; Yan, K.; Dickman, M.B.; Leslie, J.F. Electrophoretic karyotypes distinguish the biological species of Gibberella fujikuroi (Fusarium section Liseola). Mol. Plant-Microbe Interact. 1995, 8, 74–84. [Google Scholar] [CrossRef]
- Slinski, S.; Kirkpatrick, S.C.; Gordon, T.R. Inheritance of virulence in Fusarium circinatum, the cause of pitch canker in trees. Plant Pathol. 2016, 65, 1292–1296. [Google Scholar] [CrossRef]
- Fumero, M.V.; Villani, A.; Susca, A.; Haidukowski, M.; Cimmarusti, M.T.; Toomajian, C.; Leslie, J.F.; Chulze, S.N.; Moretti, A. Fumonisin and beauvericin chemotypes and genotypes of the sister species Fusarium subglutinans and Fusarium temperatum. Appl. Environ. Microbiol. 2020, 86, e00133-00120. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Statistic | FSP34 | KS17 | ||
|---|---|---|---|---|
| Previous 1 | Current | Previous 2 | Current | |
| Genome size (bp) | 43,932,912 | 45,020,843 | 46,325,048 | 44,380,849 |
| G+C content (%) | 47.41 | 47.00 | 44.69 | 47.26 |
| Number of open reading frames | 14,923 3 | 15,490 | 16,502 4 | 15,113 |
| Gene density (orfs/Mb) | 339.68 | 344.06 | 356.22 | 340.53 |
| Number of scaffolds | 585 | 49 | 6033 | 96 |
| N50 (bp) | 363,633 | 4,313,168 | 95,695 | 4,401,926 |
| Average scaffold size (bp) | 75,085 | 1,667,439 | 7679 | 1,431,640 |
| Unmapped scaffolds (total % of genome) | 418 (3.03%) | 15 (0.60%) | - 5 | 19 (0.78%) |
| Repeat content 6 | 2.81% | 8.75% | - | 8.60% |
| Genome completeness 7 | 94.8% | 97.3% | 76.2% | 98.1% |
| Pseudomolecule | FSP34 1 | KS17current | |
|---|---|---|---|
| Previous | Current | ||
| 1 | 6,190,704 (14) | 6,407,689 (2) | 6,397,914 (8) |
| 2 | 4,773,114 (22) | 5,066,197 (3) | 4,709,326 (5) |
| 3 | 4,756,822 (18) | 5,081,888 (3) | 5,148,568 (3) |
| 4 | 4,140,424 (12) | 4,313,168 (2) | 4,401,926 (5) |
| 5 | 4,399,406 (10) | 4,432,553 (2) | 4,304,443 (7) |
| 6 | 4,053,349 (21) | 4,301,895 (3) | 4,219,930 (9) |
| 7 | 3,472,423 (19) | 3,541,054 (5) | 3,312,103 (8) |
| 8 | 3,024,507 (17) | 3,172,915 (5) | 3,066,990 (9) |
| 9 | 2,773,158 (8) | 2,981,544 (1) | 2,828,005 (4) |
| 10 | 2,371,510 (16) | 2,698,820 (3) | 2,483,521 (7) |
| 11 | 2,122,486 (11) | 2,228,420 (3) | 2,291,537 (5) |
| 12 | 525,791 (6) | 525,065 (1) | 870,680 (3) |
| Content Estimates | FSP34 Pseudomolecule 12 1 | KS17 Pseudomolecule 12 2 | |||
|---|---|---|---|---|---|
| Whole Molecule | Distal | Middle | Proximal | ||
| G+C content (%) | 46.36 | 41.73 | 31.88 | 45.18 | 41.48 |
| Gene density (orfs/Mb) | 325.67 | 294.01 | 123.53 | 349.52 | 300.00 |
| Repeats (%) | 6.92 | 26.63 | 59.67 | 12.67 | 33.48 |
| Data/Properties | FSP34 | KS17 1 | References |
|---|---|---|---|
| Strain origin | |||
| Collection date | Unknown date between March 1993 and April 1995 | October 2005 | [50,52] |
| Collector | TR Gordon | OM Mashandula, ET Steenkamp | [52] |
| Host plant | Pinus radiata | Pinus radiata | [52] |
| Host tissue | Tissue from the leading edge of a canker on the branch of a mature tree | Diseased root tissue of a nursery seedling | [52] |
| Geographic location | Monterey, California (USA) | Karatara, Western Cape (South Africa) | [52] |
| Location description | Exact location is unknown, but collected in a region where mature trees of native Pinus species displayed symptoms of pitch canker | Commercial seedling nursery; collected during an outbreak of F. circinatum-associated root disease | [52] |
| Reproductive biology | |||
| Mating type | MAT1-1 | MAT1-2 | [15] (unpublished) |
| Fertility | Male fertile; mostly female sterile; also capable of mating with sexually compatible strains of Fusarium temperatum to produce fertile hybrid progeny. | Male fertile; displays some level of fertility as a female. | [53] (unpublished) |
| Source population dynamics | Forms part of a population with limited genetic diversity that propagates asexually | Forms part of a moderately diverse population that propagates mainly asexually. | [54] |
| Growth in culture | Grows at 5–30 °C, but not at 35 °C; grows faster than KS17 at 15–30 °C. | Grows at 5–30 °C, but not at 35 °C; grows slower than FSP34 at 15–30 °C. | This study |
| Pathogenicity | Capable of inducing lesions when inoculated onto the apices of the main stems of P. patula seedlings; more virulent than KS17. | Capable of inducing lesions when inoculated onto the apices of the main stems of P. patula seedlings; less virulent than FSP34. | This study |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Vos, L.; van der Nest, M.A.; Santana, Q.C.; van Wyk, S.; Leeuwendaal, K.S.; Wingfield, B.D.; Steenkamp, E.T. Chromosome-Level Assemblies for the Pine Pitch Canker Pathogen Fusarium circinatum. Pathogens 2024, 13, 70. https://doi.org/10.3390/pathogens13010070
De Vos L, van der Nest MA, Santana QC, van Wyk S, Leeuwendaal KS, Wingfield BD, Steenkamp ET. Chromosome-Level Assemblies for the Pine Pitch Canker Pathogen Fusarium circinatum. Pathogens. 2024; 13(1):70. https://doi.org/10.3390/pathogens13010070
Chicago/Turabian StyleDe Vos, Lieschen, Magriet A. van der Nest, Quentin C. Santana, Stephanie van Wyk, Kyle S. Leeuwendaal, Brenda D. Wingfield, and Emma T. Steenkamp. 2024. "Chromosome-Level Assemblies for the Pine Pitch Canker Pathogen Fusarium circinatum" Pathogens 13, no. 1: 70. https://doi.org/10.3390/pathogens13010070
APA StyleDe Vos, L., van der Nest, M. A., Santana, Q. C., van Wyk, S., Leeuwendaal, K. S., Wingfield, B. D., & Steenkamp, E. T. (2024). Chromosome-Level Assemblies for the Pine Pitch Canker Pathogen Fusarium circinatum. Pathogens, 13(1), 70. https://doi.org/10.3390/pathogens13010070