

Genetic Diversity and Population Structure of Leishmania infantum in Morocco as Revealed by Multilocus Sequence Typing (MLST) Approach

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Samples
2.3. DNA Extraction and Leishmania Species Identification
2.4. PCR Amplification of the Eight Markers
2.5. PCR Products Sequencing
2.6. Genetic Diversity and Phylogeny Analyses
2.7. Recombination Evaluation
2.8. Temporal Variation Analysis
2.9. Analysis of the Genetic Exchange between L. infantum and L. tropica
3. Results
3.1. Leishmania infantum Identification and Markers Amplification
3.2. Leishmania infantum Genetic Diversity Analyses
3.2.1. Genetic Diversity of All L. infantum Strains
3.2.2. Genetic Diversity of Canine and Human Groups
3.2.3. Genetic Diversity of the Human VL and CL Sub-Groups
3.3. Demographic Analysis and Genetic Structure
3.4. Genotypic Variation in L. infantum
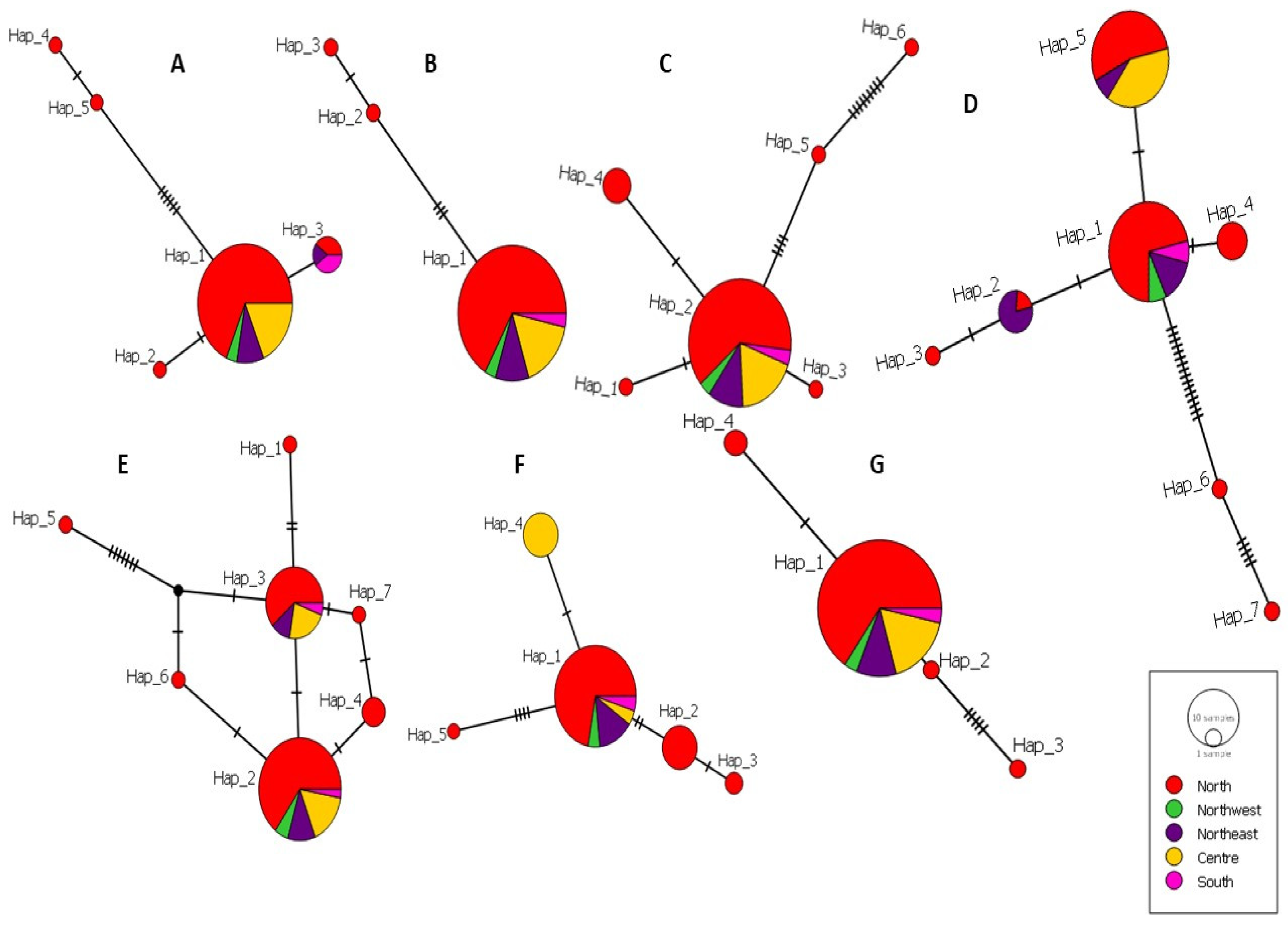
3.5. Haplotype Diversity of L. infantum
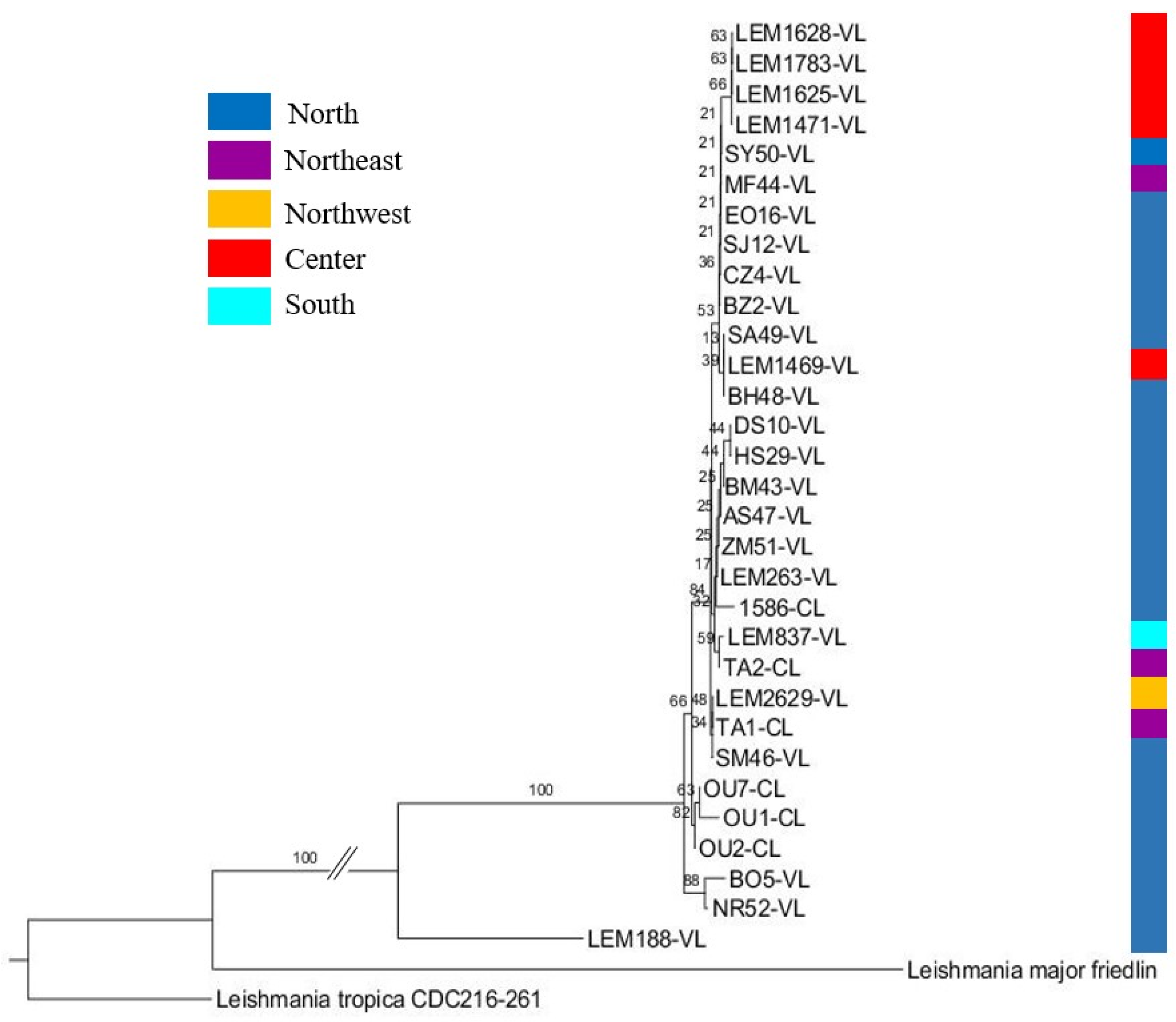
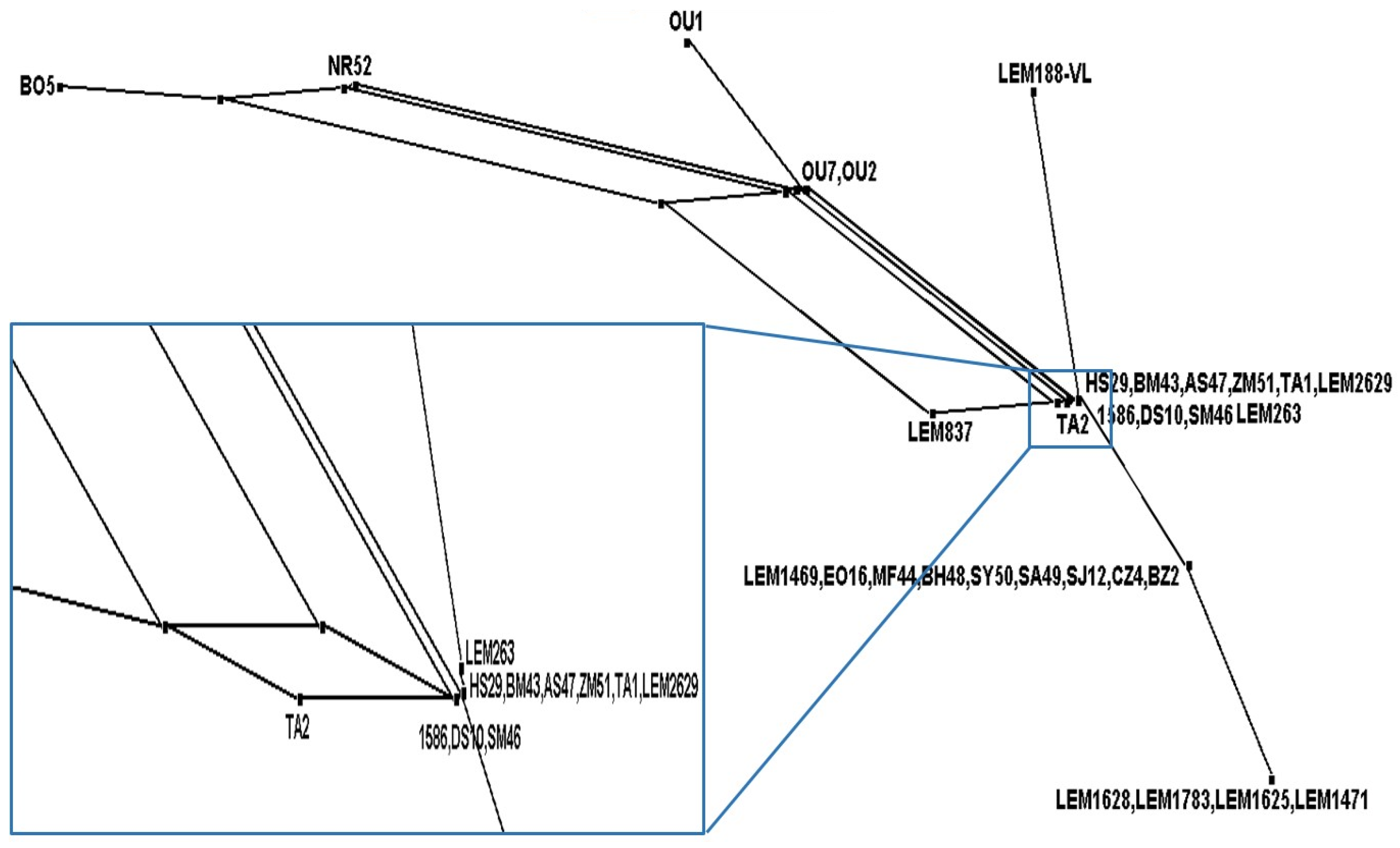
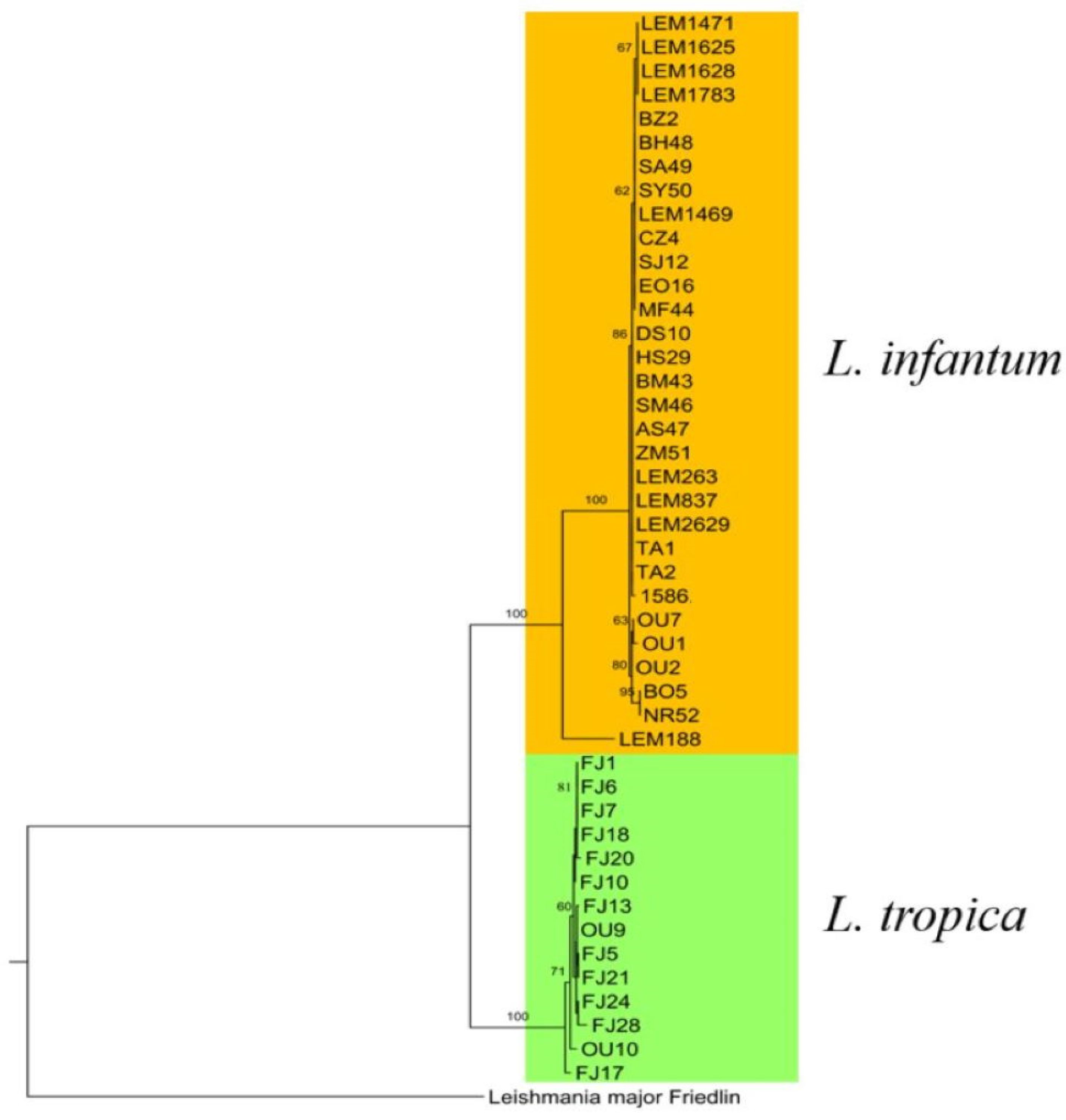
3.6. Phylogenetic Analysis of L. infantum
3.7. Recombination among L. infantum Strains
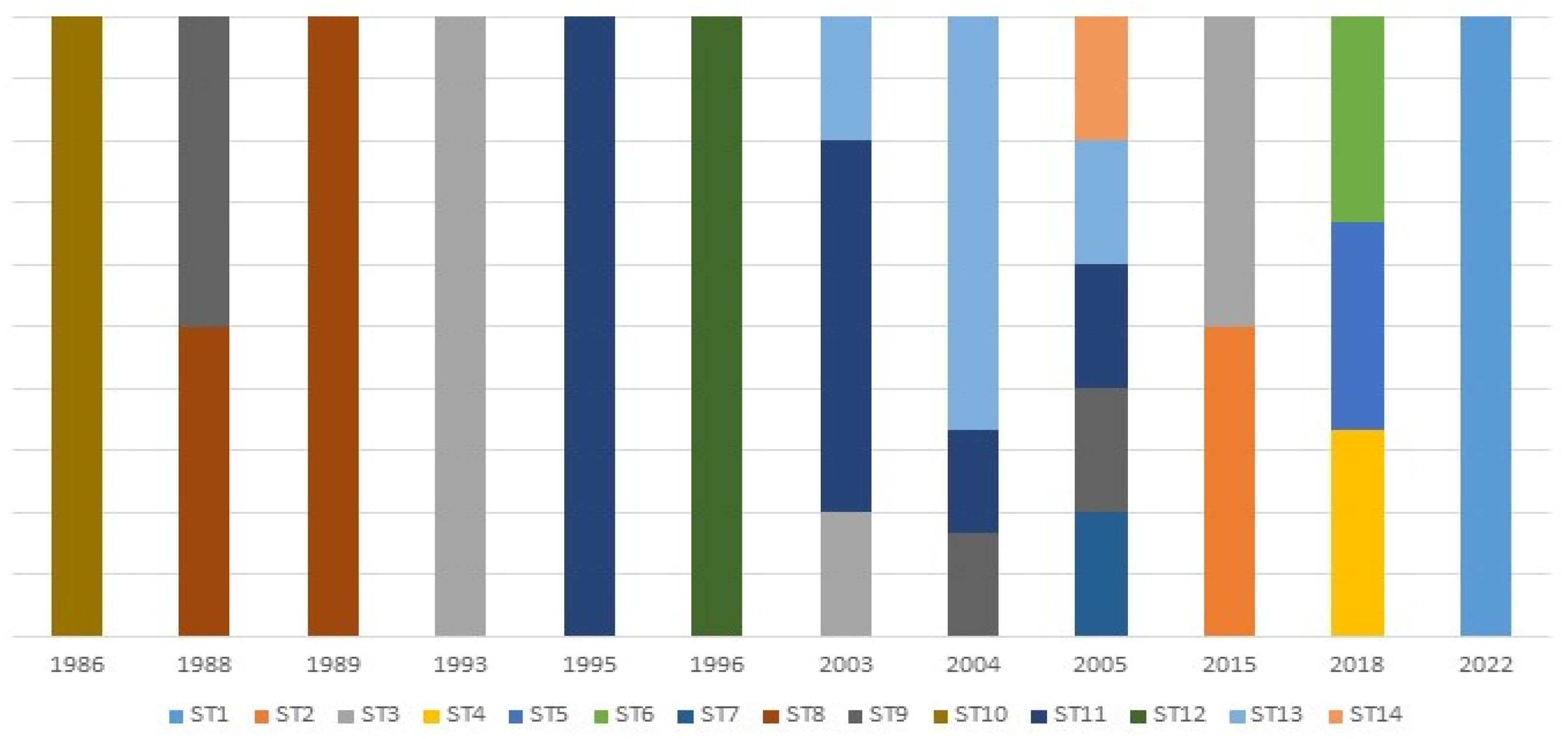
3.8. Temporal Variation Analysis
3.9. Analysis of the Genetic Exchange between L. infantum and L. tropica
3.9.1. Genotypic Variation Analysis of L. infantum and L. tropica
3.9.2. Haplotypes Diversity of L. infantum and L. tropica
3.9.3. Phylogenetic Analysis of L. infantum and L. tropica
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ashford, R.W. The leishmaniases as emerging and reemerging zoonoses. Int. J. Parasitol. 2000, 30, 1269–1281. [Google Scholar] [CrossRef]
- de Vries, H.J.; Reedijk, S.H.; Schallig, H.D. Cutaneous leishmaniasis: Recent developments in diagnosis and management. Am. J. Clin. Dermatol. 2015, 16, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Remlinger, P. Un cas de Kala-azar infantile observé au Maroc. Arch. Inst. Pasteur Afr. Noi 1921, 1, 240–241. [Google Scholar]
- Rhajaoui, M. Human leishmaniases in Morocco: A nosogeographical diversity. Pathol. Biol. 2011, 59, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Murray, H.W.; Berman, J.D.; Davies, C.R.; Saravia, N.G. Advances in leishmaniasis. Lancet 2005, 366, 1561–1577. [Google Scholar] [CrossRef] [PubMed]
- van Griensven, J.; Diro, E. Visceral leishmaniasis. Infect. Dis. Clin. N. Am. 2012, 26, 309–322. [Google Scholar] [CrossRef] [PubMed]
- El Mazini, S.; Ejghal, R.; Bekhti, K.; Lemrani, M. The Sporadic cutaneous leishmaniasis due to Leishmania infantum in Morocco: A presumably trend towards endemicity. Acta Trop. 2022, 227, 106288. [Google Scholar] [CrossRef] [PubMed]
- Hmamouch, A.; Amarir, F.; Fellah, H.; Karzaz, M.; Bekhti, K.; Rhajaoui, M.; Sebti, F. Coexistence of Leishmania tropica and Leishmania infantum in Sefrou province, Morocco. Acta Trop. 2013, 130, 94–99. [Google Scholar]
- Mhaidi, I.; El Kacem, S.; Ait Kbaich, M.; El Hamouchi, A.; Sarih, M.; Akarid, K.; Lemrani, M. Molecular identification of Leishmania infection in the most relevant sand fly species and in patient skin samples from a cutaneous leishmaniasis focus, in Morocco. PLoS Negl. Trop. Dis. 2018, 12, e0006315. [Google Scholar] [CrossRef]
- Chaffai, M.; Ben Rachid, M.S.; Ben-Ismail, R.; Ben Osman, A.; Makni, N. Clinico-epidemiologic forms of cutaneous leishmaniasis in Tunisia. Ann. Dermatol. Venereol. 1988, 115, 1255–1260. [Google Scholar]
- Rioux, J.A.; Mahjoub, J.; Gallego, M.; Dereure, J.; Perieres, J.; Lahmrani, A.J.B.S.F.P. Human cutaneous leishmaniasis due to Leishmania infantum zymodeme MON-24 in Morocco. Bull. Soc. Fr. Parasitol. 1996, 14, 179–183. [Google Scholar]
- Bachi, F. The epidemiological and clinical aspects of leishmaniasis in Algeria. La Lett. L’infectiologue 2006, 21, 9–15. [Google Scholar]
- Rhajaoui, M.; Nasereddin, A.; Fellah, H.; Azmi, K.; Amarir, F.; Al-Jawabreh, A.; Ereqat, S.; Planer, J.; Abdeen, Z. New clinico-epidemiologic profile of cutaneous leishmaniasis, Morocco. Emerg. Infect Dis. 2007, 13, 1358–1360. [Google Scholar] [CrossRef] [PubMed]
- Mouttaki, T.; Maksouri, H.; El Mabrouki, J.; Merino-Espinosa, G.; Fellah, H.; Itri, M.; Martin-Sanchez, J.; Soussi-Abdallaoui, M.; Chiheb, S.; Riyad, M. Concomitant visceral and localized cutaneous leishmaniasis in two Moroccan infants. Infect. Dis. Poverty 2018, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Nejjar, R.; Lemrani, M.; Malki, A.; Ibrahimy, S.; Amarouch, H.; Benslimane, A. Canine leishmaniasis due to Leishmania infantum MON-1 in northern Morocco. Parasite 1998, 5, 325–330. [Google Scholar] [CrossRef]
- Haralambous, C.; Dakkak, A.; Pratlong, F.; Dedet, J.P.; Soteriadou, K. First detection and genetic typing of Leishmania infantum MON-24 in a dog from the Moroccan Mediterranean coast: Genetic diversity of MON-24. Acta Trop. 2007, 103, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Echchakery, M.; Chicharro, C.; Boussaa, S.; Nieto, J.; Carrillo, E.; Sheila, O.; Moreno, J.; Boumezzough, A. Molecular detection of Leishmania infantum and Leishmania tropica in rodent species from endemic cutaneous leishmaniasis areas in Morocco. Parasit Vectors 2017, 10, 454. [Google Scholar] [CrossRef] [PubMed]
- Guessous-Idrissi, N.; Hamdani, A.; Rhalem, A.; Riyad, M.; Sahibi, H.; Dehbi, F.; Bichichi, M.; Essari, A.; Berrag, B. Epidemiology of human visceral leishmaniasis in Taounate, a northern province of Morocco. Parasite 1997, 4, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Es-Sette, N.; Ajaoud, M.; Laamrani-Idrissi, A.; Mellouki, F.; Lemrani, M. Molecular detection and identification of Leishmania infection in naturally infected sand flies in a focus of cutaneous leishmaniasis in northern Morocco. Parasit Vectors 2014, 7, 305. [Google Scholar] [CrossRef]
- Schönian, G.; Mauricio, I.; Gramiccia, M.; Cañavate, C.; Boelaert, M.; Dujardin, J.C. Leishmaniases in the Mediterranean in the era of molecular epidemiology. Trends Parasitol. 2008, 24, 135–142. [Google Scholar] [CrossRef]
- Ochsenreither, S.; Kuhls, K.; Schaar, M.; Presber, W.; Schönian, G. Multilocus microsatellite typing as a new tool for discrimination of Leishmania infantum MON-1 strains. J. Clin. Microbiol. 2006, 44, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Schönian, G.; Kuhls, K.; Mauricio, I.L. Molecular approaches for a better understanding of the epidemiology and population genetics of Leishmania. Parasitology 2011, 138, 405–425. [Google Scholar] [CrossRef]
- Lauthier, J.J.; Ruybal, P.; Barroso, P.A.; Hashiguchi, Y.; Marco, J.D.; Korenaga, M. Development of a Multilocus sequence typing (MLST) scheme for Pan-Leishmania. Acta Trop. 2020, 201, 105189. [Google Scholar] [CrossRef] [PubMed]
- Marco, J.D.; Barroso, P.A.; Locatelli, F.M.; Cajal, S.P.; Hoyos, C.L.; Nevot, M.C.; Lauthier, J.J.; Tomasini, N.; Juarez, M.; Estévez, J.O.; et al. Multilocus sequence typing approach for a broader range of species of Leishmania genus: Describing parasite diversity in Argentina. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2015, 30, 308–317. [Google Scholar] [CrossRef] [PubMed]
- El Hamouchi, A.; El Kacem, S.; Ejghal, R.; Lemrani, M. Genetic polymorphism in Leishmania infantum isolates from human and animals determined by nagt PCR-RFLP. Infect. Dis. Poverty 2018, 7, 54. [Google Scholar] [CrossRef] [PubMed]
- Mauricio, I.L.; Yeo, M.; Baghaei, M.; Doto, D.; Pratlong, F.; Zemanova, E.; Dedet, J.P.; Lukes, J.; Miles, M.A. Towards multilocus sequence typing of the Leishmania donovani complex: Resolving genotypes and haplotypes for five polymorphic metabolic enzymes (ASAT, GPI, NH1, NH2, PGD). Int. J. Parasitol. 2006, 36, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Zemanová, E.; Jirků, M.; Mauricio, I.L.; Horák, A.; Miles, M.A.; Lukes, J. The Leishmania donovani complex: Genotypes of five metabolic enzymes (ICD, ME, MPI, G6PDH, and FH), new targets for multilocus sequence typing. Int. J. Parasitol. 2007, 37, 149–160. [Google Scholar] [CrossRef]
- El Kacem, S.; Kbaich, M.A.; Daoui, O.; Charoute, H.; Mhaidi, I.; Ejghal, R.; Barhoumim, M.; Guizani, I.; Bennani, H.; Lemrani, M. Multilocus sequence analysis provides new insight into population structure and genetic diversity of Leishmania tropica in Morocco. Infect. Genet. Evol. 2021, 93, 104932. [Google Scholar] [CrossRef]
- Schönian, G.; Schweynoch, C.; Zlateva, K.; Oskam, L.; Kroon, N.; Gräser, Y.; Presber, W. Identification and determination of the relationships of species and strains within the genus Leishmania using single primers in the polymerase chain reaction. Mol. Biochem. Parasitol. 1996, 77, 19–29. [Google Scholar] [CrossRef]
- Schönian, G.; Nasereddin, A.; Dinse, N.; Schweynoch, C.; Schallig, H.D.; Presber, W.; Jaffe, C.L. PCR diagnosis and characterization of Leishmania in local and imported clinical samples. Diagn. Microbiol. Infect. Dis. 2003, 47, 349–358. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Lischer, H.E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Tomasini, N.; Lauthier, J.J.; Llewellyn, M.S.; Diosque, P. MLSTest: Novel software for multi-locus sequence data analysis in eukaryotic organisms. Infect. Genet. Evol. 2013, 20, 188–196. [Google Scholar] [CrossRef]
- Stöver, B.C.; Müller, K.F. TreeGraph 2: Combining and visualizing evidence from different phylogenetic analyses. BMC Bioinform. 2010, 11, 7. [Google Scholar] [CrossRef]
- Leigh, J.; Bryant, D. PopART: Full-feature software for haplotype network construction. Methods Ecol. Evol 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Hudson, R.R.; Kaplan, N.L. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 1985, 111, 147–164. [Google Scholar] [CrossRef]
- Hudson, R.R. Estimating the recombination parameter of a finite population model without selection. Genet. Res. 1987, 50, 245–250. [Google Scholar] [CrossRef]
- El Hamouchi, A.; Ejghal, R.; Hida, M.; Lemrani, M. Intraspecific genetic variability in a population of Moroccan Leishmania infantum revealed by PCR-RFLP of kDNA minicircles. Acta Trop. 2017, 169, 142–149. [Google Scholar] [CrossRef]
- Gállego, M.; Pratlong, F.; Fisa, R.; Riera, C.; Rioux, J.A.; Dedet, J.P.; Portús, M. The life-cycle of Leishmania infantum MON-77 in the Priorat (Catalonia, Spain) involves humans, dogs and sandflies; also literature review of distribution and hosts of L. infantum zymodemes in the Old World. Trans. R. Soc. Trop. Med. Hyg. 2001, 95, 269–271. [Google Scholar] [CrossRef] [PubMed]
- WHO. Leishmaniasis 2023. Available online: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed on 23 June 2022).
- Bennasar, A.; Mulet, M.; Lalucat, J.; García-Valdés, E. PseudoMLSA: A database for multigenic sequence analysis of Pseudomonas species. BMC Microbiol. 2010, 10, 118. [Google Scholar] [CrossRef] [PubMed]
- Weirather, J.L.; Jeronimo, S.M.; Gautam, S.; Sundar, S.; Kang, M.; Kurtz, M.A.; Haque, R.; Schriefer, A.; Talhari, S.; Carvalho, E.M.; et al. Serial quantitative PCR assay for detection, species discrimination, and quantification of Leishmania spp. in human samples. J. Clin. Microbiol. 2011, 49, 3892–3904. [Google Scholar] [CrossRef] [PubMed]
- Boité, M.C.; Mauricio, I.L.; Miles, M.A.; Cupolillo, E. New insights on taxonomy, phylogeny and population genetics of Leishmania (Viannia) parasites based on multilocus sequence analysis. PLoS Negl. Trop. Dis. 2012, 6, e1888. [Google Scholar] [CrossRef]
- Francisco, A.P.; Bugalho, M.; Ramirez, M.; Carriço, J.A. Global optimal eBURST analysis of multilocus typing data using a graphic matroid approach. BMC Bioinform. 2009, 10, 152. [Google Scholar] [CrossRef]
- Ortuño, M.; Latrofa, M.S.; Iborra, M.A.; Pérez-Cutillas, P.; Bernal, L.J.; Risueño, J.; Muñoz, C.; Bernal, A.; Sánchez-Lopez, P.F.; Segovia, M.; et al. Genetic diversity and phylogenetic relationships between Leishmania infantum from dogs, humans and wildlife in south-east Spain. Zoonoses Public Health 2019, 66, 961–973. [Google Scholar] [CrossRef]
- Amro, A.; Hamdi, S.; Lemrani, M.; Mouna, I.; Mohammed, H.; Mostafa, S.; Rhajaoui, M.; Hamarsheh, O.; Schönian, G. Moroccan Leishmania infantum: Genetic diversity and population structure as revealed by multi-locus microsatellite typing. PLoS ONE 2013, 8, e77778. [Google Scholar] [CrossRef]
- Pomares, C.; Marty, P.; Bañuls, A.L.; Lemichez, E.; Pratlong, F.; Faucher, B.; Jeddi, F.; Moore, S.; Michel, G.; Aluru, S.; et al. Genetic Diversity and Population Structure of Leishmania infantum from Southeastern France: Evaluation Using Multi-Locus Microsatellite Typing. PLoS Negl. Trop. Dis. 2016, 10, e0004303. [Google Scholar] [CrossRef]
- Banu, S.S.; Meyer, W.; Ferreira-Paim, K.; Wang, Q.; Kuhls, K.; Cupolillo, E.; Schönian, G.; Lee, R. A novel multilocus sequence typing scheme identifying genetic diversity amongst Leishmania donovani isolates from a genetically homogeneous population in the Indian subcontinent. Int. J. Parasitol. 2019, 49, 555–567. [Google Scholar] [CrossRef]
- Hunter, P.R.; Gaston, M.A. Numerical index of the discriminatory ability of typing systems: An application of Simpson’s index of diversity. J. Clin. Microbiol. 1988, 26, 2465–2466. [Google Scholar] [CrossRef] [PubMed]
- Botilde, Y.; Laurent, T.; Quispe Tintaya, W.; Chicharro, C.; Cañavate, C.; Cruz, I.; Kuhls, K.; Schönian, G.; Dujardin, J.C. Comparison of molecular markers for strain typing of Leishmania infantum. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2006, 6, 440–446. [Google Scholar] [CrossRef]
- Chargui, N.; Amro, A.; Haouas, N.; Schönian, G.; Babba, H.; Schmidt, S.; Ravel, C.; Lefebvre, M.; Bastien, P.; Chaker, E.; et al. Population structure of Tunisian Leishmania infantum and evidence for the existence of hybrids and gene flow between genetically different populations. Int. J. Parasitol. 2009, 39, 801–811. [Google Scholar] [CrossRef]
- Ferroglio, E.; Romano, A.; Trisciuoglio, A.; Poggi, M.; Ghiggi, E.; Sacchi, P.; Biglino, A. Characterization of Leishmania infantum strains in blood samples from infected dogs and humans by PCR-RFLP. Trans. R. Soc. Trop. Med. Hyg. 2006, 100, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.Z.; Nakao, R.; Sakurai, T.; Kato, H.; Qu, J.Q.; Chai, J.J.; Chang, K.P.; Schönian, G.; Katakura, K. Genetic diversity of Leishmania donovani/infantum complex in China through microsatellite analysis. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2014, 22, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Kuhls, K.; Chicharro, C.; Cañavate, C.; Cortes, S.; Campino, L.; Haralambous, C.; Soteriadou, K.; Pratlong, F.; Dedet, J.P.; Mauricio, I.; et al. Differentiation and gene flow among European populations of Leishmania infantum MON-1. PLoS Negl. Trop. Dis. 2008, 2, e261. [Google Scholar] [CrossRef]
- Seridi, N.; Amro, A.; Kuhls, K.; Belkaid, M.; Zidane, C.; Al-Jawabreh, A.; Schönian, G. Genetic polymorphism of Algerian Leishmania infantum strains revealed by multilocus microsatellite analysis. Microbes Infect. 2008, 10, 1309–1315. [Google Scholar] [CrossRef]
- Alvar, J.; Jiménez, M. Could infected drug-users be potential Leishmania infantum reservoirs? AIDS 1994, 8, 854. [Google Scholar] [CrossRef]
- Rossi, M.; Fasel, N. How to master the host immune system? Leishmania parasites have the solutions! Int. Immunol. 2018, 30, 103–111. [Google Scholar] [CrossRef]
- Hakkour, M.; Hmamouch, A.; El Alem, M.M.; Rhalem, A.; Amarir, F.; Touzani, M.; Sadak, A.; Fellah, H.; Sebti, F. New epidemiological aspects of visceral and cutaneous leishmaniasis in Taza, Morocco. Parasites Vectors 2016, 9, 612. [Google Scholar] [CrossRef][Green Version]
- Kuhls, K.; Alam, M.Z.; Cupolillo, E.; Ferreira, G.E.; Mauricio, I.L.; Oddone, R.; Feliciangeli, M.D.; Wirth, T.; Miles, M.A.; Schönian, G. Comparative microsatellite typing of new world Leishmania infantum reveals low heterogeneity among populations and its recent old world origin. PLoS Negl. Trop. Dis. 2011, 5, e1155. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, Y.; Wang, J.; Bao, Y.; Guan, L.; Gao, C.; Shi, F. Molecular characterization of Leishamania isolates from China by inter-simple sequence repeat polymerase chain reaction. Parasitol. Res. 2010, 106, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Belli, A.; García, D.; Palacios, X.; Rodriguez, B.; Valle, S.; Videa, E.; Tinoco, E.; Marín, F.; Harris, E. Widespread atypical cutaneous Leishmaniasis caused by Leishmania (L.) chagasi in Nicaragua. Am. J. Trop. Med. Hyg. 1999, 61, 380–385. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Noyes, H.; Chance, M.; Ponce, C.; Ponce, E.; Maingon, R. Leishmania chagasi: Genotypically similar parasites from Honduras cause both visceral and cutaneous leishmaniasis in humans. Exp. Parasitol. 1997, 85, 264–273. [Google Scholar] [CrossRef]
- Cruz, I.; Cañavate, C.; Rubio, J.M.; Morales, M.A.; Chicharro, C.; Laguna, F.; Jiménez-Mejías, M.; Sirera, G.; Videla, S.; Alvar, J. A nested polymerase chain reaction (Ln-PCR) for diagnosing and monitoring Leishmania infantum infection in patients co-infected with human immunodeficiency virus. Trans. R. Soc. Trop. Med. Hyg. 2002, 96 (Suppl. S1), S185–S189. [Google Scholar] [CrossRef]
- Alvar, J.; Cañavate, C.; Gutiérrez-Solar, B.; Jiménez, M.; Laguna, F.; López-Vélez, R.; Molina, R.; Moreno, J. Leishmania and human immunodeficiency virus coinfection: The first 10 years. Clin. Microbiol. Rev. 1997, 10, 298–319. [Google Scholar] [CrossRef]
- Pratlong, F.; Reynes, J.; Dereure, J.; Baixench, M.T.; Marchou, B.; Lefebvre, M.; Janbon, F. Characterization of Leishmania isolates from two AIDS patients originating from Valencia, Spain. Trans. R. Soc. Trop. Med. Hyg. 1993, 87, 705–706. [Google Scholar] [CrossRef]
- Jiménez, M.I.; Laguna, F.; de la Torré, F.; Solís, F.; Pratlong, F.; Alvar, J. New Leishmania (Leishmania) infantum zymodemes responsible for visceral leishmaniasis in patients co-infected with HIV in Spain. Trans. R. Soc. Trop. Med. Hyg. 1995, 89, 33. [Google Scholar] [CrossRef]
- Momen, H.; Pacheco, R.S.; Cupolillo, E.; Grimaldi Júnior, G. Molecular evidence for the importation of Old World Leishmania into the Americas. Biol. Res. 1993, 26, 249–255. [Google Scholar]
- Marlow, M.A.; Boité, M.C.; Ferreira, G.E.; Steindel, M.; Cupolillo, E. Multilocus sequence analysis for Leishmania braziliensis outbreak investigation. PLoS Negl. Trop. Dis. 2014, 8, e2695. [Google Scholar] [CrossRef]
- Ghatee, M.A.; Mirhendi, H.; Karamian, M.; Taylor, W.R.; Sharifi, I.; Hosseinzadeh, M.; Kanannejad, Z. Population structures of Leishmania infantum and Leishmania tropica the causative agents of kala-azar in Southwest Iran. Parasitol. Res. 2018, 117, 3447–3458. [Google Scholar] [CrossRef]
- Kelly, J.M.; Law, J.M.; Chapman, C.J.; Van Eys, G.J.; Evans, D.A. Evidence of genetic recombination in Leishmania. Mol. Biochem. Parasitol. 1991, 46, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Nolder, D.; Roncal, N.; Davies, C.R.; Llanos-Cuentas, A.; Miles, M.A. Multiple hybrid genotypes of Leishmania (Viannia) in a focus of mucocutaneous Leishmaniasis. Am. J. Trop. Med. Hyg. 2007, 76, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Odiwuor, S.; De Doncker, S.; Maes, I.; Dujardin, J.C.; Van der Auwera, G. Natural Leishmania donovani/Leishmania aethiopica hybrids identified from Ethiopia. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2011, 11, 2113–2118. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.B.; Downing, T.; Smith, B.A.; Imamura, H.; Sanders, M.; Svobodova, M.; Volf, P. Genomic confirmation of hybridisation and recent inbreeding in a vector-isolated Leishmania population. PLoS Genet. 2014, 10, e1004092. [Google Scholar] [CrossRef] [PubMed]
- El Baidouri, F.; Diancourt, L.; Berry, V.; Chevenet, F.; Pratlong, F.; Marty, P.; Ravel, C. Genetic structure and evolution of the Leishmania genus in Africa and Eurasia: What does MLSA tell us. PLoS Negl. Trop. Dis. 2013, 7, e2255. [Google Scholar] [CrossRef]
- Marty, P.; Le Fichoux, Y.; Pratlong, F.; Rioux, J.A.; Rostain, G.; Lacour, J.P. Cutaneous leishmaniasis due to Leishmania tropica in a young Moroccan child observed in Nice, France. Trans. R. Soc. Trop. Med. Hyg. 1989, 83, 510. [Google Scholar] [CrossRef]
- El Miri, H.; Faraj, C.; Himmi, O.; Hmamouch, A.; Maniar, S.; Laaroussi, T.; Rhajaoui, M.; Sebti, F.; Benhoussa, A. Cutaneous leishmaniasis in Ouazzane and Sidi Kacem provinces, Morocco (1997–2012). Bull. Soc. Pathol. Exot. 2016, 109, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Herrera, G.; Hernández, C.; Ayala, M.S.; Flórez, C.; Teherán, A.A.; Ramírez, J.D. Evaluation of a Multilocus Sequence Typing (MLST) scheme for Leishmania (Viannia) braziliensis and Leishmania (Viannia) panamensis in Colombia. Parasites Vectors 2017, 10, 236. [Google Scholar] [CrossRef]
- Gouzelou, E.; Haralambous, C.; Antoniou, M.; Christodoulou, V.; Martinković, F.; Živičnjak, T.; Smirlis, D.; Pratlong, F.; Dedet, J.P.; Özbel, Y.; et al. Genetic diversity and structure in Leishmania infantum populations from southeastern Europe revealed by microsatellite analysis. Parasites Vectors 2013, 6, 342. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Leishmania Species | Disease Form | Strain Code | Geographic Origin | Sequence Types | Haplotypes | ||||||
| alat | fh | g6pd | gpi | me | pgd | pgm | |||||
| L. infantum | HIV+/CL | 1586 | North | ST1 | 1–2 | 1 | 1–2 | 1–2 | 1 | 1 | 1 |
| CL | TA2 | Northeast | ST2 | 1–3 | 1 | 2 | 2 | 1 | 1 | 1 | |
| CL | TA1 | Northeast | ST3 | 1 | 1 | 2 | 2–3 | 1 | 1 | 1 | |
| CL | OU1 | North | ST4 | 1 | 1 | 2 | 2 | 2–3 | 1 | 1 | |
| CL | OU2 | North | ST5 | 1 | 1 | 2 | 2 | 2 | 1 | 1 | |
| CL | OU7 | North | ST6 | 1 | 1 | 2–3 | 2 | 2–3 | 2–3 | 1 | |
| VL | NR52 | North | ST7 | 1 | 1 | 4 | 4 | 2 | 4 | 1 | |
| Canine | LEM2629 | Northwest | ST3 | 1 | 1 | 2 | 2 | 1 | 1 | 1 | |
| Canine | LEM1783 | Center | ST8 | 1 | 1 | 2 | 2–3 | 4 | 5 | 1 | |
| Canine | LEM1628 | Center | ST8 | 1 | 1 | 2 | 2–3 | 4 | 5 | 1 | |
| Canine | LEM1625 | Center | ST8 | 1 | 1 | 2 | 2–3 | 4 | 5 | 1 | |
| Canine | LEM1471 | Center | ST8 | 1 | 1 | 2 | 2–3 | 4 | 5 | 1 | |
| Canine | LEM1469 | Center | ST9 | 1 | 1 | 2 | 2 | 1 | 5 | 1 | |
| Canine | LEM837 | South | ST10 | 3 | 1 | 2 | 2–3 | 1 | 1 | 1 | |
| Canine | LEM263 | North | ST11 | 1 | 1 | 2 | 2–3 | 1 | 1 | 1 | |
| Canine | LEM188 | North | ST12 | 4–5 | 2–3 | 5–6 | 5–6 | 1–5 | 6–7 | 2–3 | |
| VL | ZM51 | North | ST11 | 1 | 1 | 2 | 2–3 | 1 | 1 | 1 | |
| VL | SY50 | North | ST13 | 1 | 1 | 2 | 2–3 | 1 | 5 | 1 | |
| VL | SA49 | North | ST9 | 1 | 1 | 2 | 2 | 1 | 5 | 1 | |
| VL | BH48 | North | ST9 | 1 | 1 | 2 | 2 | 1 | 5 | 1 | |
| VL | AS47 | North | ST11 | 1 | 1 | 2 | 2–3 | 1 | 1 | 1 | |
| VL | SM46 | North | ST3 | 1 | 1 | 2 | 2 | 1 | 1 | 1 | |
| VL | MF44 | Northeast | ST13 | 1 | 1 | 2 | 2–3 | 1 | 5 | 1 | |
| VL | BM43 | North | ST11 | 1 | 1 | 2 | 2–3 | 1 | 1 | 1 | |
| VL | HS29 | North | ST11 | 1 | 1 | 2 | 2–3 | 1 | 1 | 1 | |
| VL | EO16 | North | ST13 | 1 | 1 | 2 | 2–3 | 1 | 5 | 1 | |
| VL | SJ12 | North | ST13 | 1 | 1 | 2 | 2–3 | 1 | 5 | 1 | |
| VL | DS10 | North | ST11 | 1 | 1 | 2 | 2–3 | 1 | 1 | 1 | |
| VL | BO5 | North | ST14 | 3 | 1 | 4 | 4–7 | 2 | 4 | 4 | |
| VL | CZ4 | North | ST13 | 1 | 1 | 2 | 2–3 | 1 | 5 | 1 | |
| VL | BZ2 | North | ST13 | 1 | 1 | 2 | 2–3 | 1 | 5 | 1 | |
| L. tropica | CL | OU9 | North | ST10 | 5 | 4–5 | 6 | 6 | 6 | 8 | 5 |
| CL | OU10 | North | ST11 | 5 | 6 | 7–8 | 6 | 6 | 8 | 6 | |
| CL | FJ1 | Center | 16 | 5 | 4 | 6 | 6 | 6 | 8–9 | 5 | |
| CL | FJ5 | Center | 14 | 5 | 4–8 | 6 | 6 | 6 | 8 | 5 | |
| CL | FJ6 | Center | 16 | 5 | 4 | 6 | 6 | 6 | 8–9 | 5 | |
| CL | FJ7 | Center | 16 | 5 | 4 | 6 | 6 | 6 | 8–9 | 5 | |
| CL | FJ10 | Center | 18 | 5–6 | 4–7 | 9–6 | 6 | 6 | 8 | 5 | |
| CL | FJ13 | Center | 19 | 5 | 4 | 6 | 6 | 6 | 9–11 | 5 | |
| CL | FJ17 | Center | 17 | 5 | 4 | 6 | 6–7 | 6–7 | 8–10 | 9 | |
| CL | FJ18 | Center | 16 | 5 | 4 | 6 | 6 | 6 | 8–9 | 5 | |
| CL | FJ20 | Center | 15 | 5 | 4–9 | 6 | 6 | 6 | 8–9 | 5 | |
| CL | FJ21 | Center | 14 | 5 | 4–8 | 6 | 6 | 6 | 8–9 | 5 | |
| CL | FJ24 | Center | 13 | 5 | 4 | 6 | 6 | 6 | 8 | 6 | |
| CL | FJ28 | Center | 12 | 5 | 5–7 | 6 | 6 | 6 | 8 | 7–8 | |
| DNA Marker | Chromosome | Primers Sequences (5′ to 3′) | Amplicon Size (bp) |
|---|---|---|---|
| alat | 12 | FW: GTGTGCATCAACCCMGGGAA RE: CGTTCAGCTCCTCGTTCCGC | 589 |
| fh | 29 | FW: GTCATCGACGAYRATGAACG RE: CAACAAGARCGGCATYTACA | 604 |
| gpi | 12 | FW: TCCAAGTCRCAYATCAACGA RE: GCATTCGTCAACGTTTCTTG | 500 |
| g6pd | 34 | FW: GATYCGMGAGAAGGAGAATG RE: CGGTCGTTGTTGATGTTGAG | 684 |
| me | 24 | FW: AYCAGGTGGARCGGTACTGG RE: TGTGGAARCCAGCAGCKATG | 687 |
| pgd | 35 | FW: TGTGAGCHTGGCRAGAATCT RE: GTATCACAACGCTGGGGAGT | 697 |
| Pgm | 21 | FW: CAGAGAAGCTGACGTCCCG RE: GACGGGTTCACGAAGAAGG | 529 |
| Cytb | Kinetoplast Maxicircle | FW: AGCGGAGAGRARAGAAAG RE: GYTCRCAATAAAATGCAAC | 618 |
| Marker | Accession Numbers |
|---|---|
| fh | MW411684.1,MW411686.1-MW411692.1,MW411694.1-MW411696.1,MW411703.1 |
| gpi | MW411726.1,MW411728.1-MW411734.1,MW411736.1-MW411738.1 |
| g6pd | MW411705.1,MW411707.1-MW411713.1,MW411715.1-MW411717.1,MW411723.1 |
| me | MW411746.1,MW411748.1-MW411754.1,MW411756.1-MW411758.1,MW411765.1 |
| pgd | MW411768.1,MW411770.1-MW411776.1,MW411778.1-MW411780.1MW411787.1 |
| Marker | Fragment Size | S | H | Hd | Pi | K | dN/dS |
|---|---|---|---|---|---|---|---|
| alat | 412 | 9 | 5 | 0.23797 | 0.00145 | 0.596 | 0.5 |
| fh | 483 | 4 | 3 | 0.06399 | 0.00046 | 0.223 | 1 |
| g6pd | 594 | 16 | 6 | 0.24008 | 0.00123 | 0.731 | 0.333 |
| gpi | 447 | 12 | 7 | 0.56531 | 0.00208 | 0.932 | 0.333 |
| me | 485 | 8 | 5 | 0.49233 | 0.002 | 0.97 | 0.333 |
| pgd | 534 | 25 | 7 | 0.62507 | 0.00354 | 1.89 | 0.4705 |
| pgm | 431 | 8 | 4 | 0.12533 | 0.00082 | 0.352 | 0.1428 |
| Concatenated | 3368 | 82 | 18 | 0.89212 | 0.00168 | 5.694 | - |
| Marker | alat | fh | g6pd | gpi | me | pgd | pgm | Concatenated | |
|---|---|---|---|---|---|---|---|---|---|
| Canine strains | S | 8 | 4 | 13 | 9 | 5 | 22 | 7 | 68 |
| H | 4 | 3 | 3 | 4 | 3 | 4 | 3 | 9 | |
| Hd | 0.398 | 0.216 | 0.215 | 0.608 | 0.582 | 0.608 | 0.216 | 0.895 | |
| Pi | 0.00382 | 0.00153 | 0.00309 | 0.00333 | 0.00199 | 0.00829 | 0.00226 | 0.00355 | |
| K | 1.575 | 0.738 | 1.836 | 1.49 | 0.967 | 4.425 | 0.974 | 12.006 | |
| Human strains | S | 2 | 0 | 1 | 4 | 3 | 4 | 1 | 17 |
| H | 3 | 1 | 2 | 5 | 3 | 5 | 2 | 13 | |
| Hd | 0.17230 | 0 | 0.16913 | 0.5563 | 0.37632 | 0.62262 | 0.08879 | 0.86364 | |
| Pi | 0.00043 | 0 | 0.00035 | 0.00157 | 0.00167 | 0.00145 | 0.00021 | 0.00083 | |
| K | 0.17548 | 0 | 0.16913 | 0.70402 | 0.80761 | 0.77696 | 0.08879 | 2.8129 | |
| Marker | alat | fh | g6pd | gpi | me | pgd | pgm | Concatenated | |
|---|---|---|---|---|---|---|---|---|---|
| VL human strains | S | 1 | 0 | 1 | 2 | 2 | 2 | 1 | 9 |
| H | 2 | 1 | 2 | 4 | 2 | 3 | 2 | 7 | |
| Hd | 0.121 | 0 | 0.226 | 0.608 | 0.299 | 0.613 | 0.121 | 0.814 | |
| Pi | 0.00029 | 0 | 0.00038 | 0.00159 | 0.00123 | 0.00139 | 0.00028 | 0.00070 | |
| K | 0.121 | 0 | 0.226 | 0.709 | 0.598 | 0.742 | 0.121 | 2.371 | |
| CL Human strains | S | 2 | 0 | 2 | 3 | 3 | 2 | 0 | 12 |
| H | 3 | 1 | 3 | 3 | 3 | 3 | 1 | 7 | |
| Hd | 0.318 | 0 | 0.318 | 0.318 | 0.666 | 0.318 | 0 | 0.864 | |
| Pi | 0.00081 | 0 | 0.00056 | 0.00142 | 0.00287 | 0.00088 | 0 | 0.00094 | |
| K | 0.333 | 0 | 0.333 | 0.636 | 1.394 | 0.469 | 0 | 3.167 | |
| All Strains | Canine Strains | Human Strains | ||
| −0.89377 | ||||
| CL strains | VL strains | |||
| Tajima’s D statistic | −2.31766 ** | −1.63404 | −0.85315 | 0.18667 |
| Among (VL, CL and CanL) | ||
|---|---|---|
| Among Populations | Within Populations | |
| d.f.* | 2 | 59 |
| Sum of Squares | 15.012 | 104.472 |
| Variance Components | 0.30236 | 1.77072 |
| Percentage Variation | 14.58% | 85.41% |
| Fst | 0.14585 | |
| p-value | <0.0001 | |
| North | Northeast | Center | |
|---|---|---|---|
| North | - | 0.0117 | 0.226 |
| Northeast | - | - | 0.455 |
| Marker | Number of Alleles | Number of Polymorphisms | Typing Efficiency | Discrimination Power (95% CI) * |
|---|---|---|---|---|
| alat | 5 | 9 | 0.556 | 0.299 (0.071–0.527) |
| fh | 2 | 4 | 0.5 | 0.065 (0–0.194) |
| g6pd | 5 | 16 | 0.312 | 0.299 (0.071–0.527) |
| gpi | 6 | 12 | 0.5 | 0.594 (0.439–0.748) |
| me | 5 | 8 | 0.625 | 0.527 (0.315–0.739) |
| pgd | 5 | 25 | 0.2 | 0.634 (0.532–0.737) |
| pgm | 3 | 8 | 0.375 | 0.127 (0–0.3) |
| Total | 14 | 82 | 0.378 | 0.91 (0.858–0.962) |
| Number of Alleles | Number of Polymorphisms | Typing Efficiency | Discrimination Power (95% CI) * | |
|---|---|---|---|---|
| alat | 5 | 24 | 0.208 | 0.511 (0.382–0.64) |
| fh | 9 | 16 | 0.562 | 0.538 (0.364–0.712) |
| g6pd | 8 | 29 | 0.276 | 0.598 (0.45–0.746) |
| gpi | 6 | 18 | 0.333 | 0.566 (0.439–0.693) |
| me | 7 | 16 | 0.438 | 0.699 (0.596–0.802) |
| pgd | 9 | 41 | 0.22 | 0.797 (0.731–0.862) |
| pgm | 7 | 18 | 0.389 | 0.543 (0.39–0.697) |
| Total | 22 | 162 | 0.315 | 0.907 (0.849–0.966) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Mazini, S.; Barhoumi, M.; Mhaidi, I.; Daoui, O.; Kbaich, M.A.; El Kacem, S.; El idrissi Saik, I.; Riyad, M.; Bekhti, K.; Guizani, I.; et al. Genetic Diversity and Population Structure of Leishmania infantum in Morocco as Revealed by Multilocus Sequence Typing (MLST) Approach. Pathogens 2023, 12, 785. https://doi.org/10.3390/pathogens12060785
El Mazini S, Barhoumi M, Mhaidi I, Daoui O, Kbaich MA, El Kacem S, El idrissi Saik I, Riyad M, Bekhti K, Guizani I, et al. Genetic Diversity and Population Structure of Leishmania infantum in Morocco as Revealed by Multilocus Sequence Typing (MLST) Approach. Pathogens. 2023; 12(6):785. https://doi.org/10.3390/pathogens12060785
Chicago/Turabian StyleEl Mazini, Sara, Mourad Barhoumi, Idris Mhaidi, Othmane Daoui, Mouad Ait Kbaich, Sofia El Kacem, Imane El idrissi Saik, Myriam Riyad, Khadija Bekhti, Ikram Guizani, and et al. 2023. "Genetic Diversity and Population Structure of Leishmania infantum in Morocco as Revealed by Multilocus Sequence Typing (MLST) Approach" Pathogens 12, no. 6: 785. https://doi.org/10.3390/pathogens12060785
APA StyleEl Mazini, S., Barhoumi, M., Mhaidi, I., Daoui, O., Kbaich, M. A., El Kacem, S., El idrissi Saik, I., Riyad, M., Bekhti, K., Guizani, I., & Lemrani, M. (2023). Genetic Diversity and Population Structure of Leishmania infantum in Morocco as Revealed by Multilocus Sequence Typing (MLST) Approach. Pathogens, 12(6), 785. https://doi.org/10.3390/pathogens12060785