
Fantastic AAV Gene Therapy Vectors and How to Find Them—Random Diversification, Rational Design and Machine Learning


{kind=link}
{kind=link}
Abstract
:1. Introduction
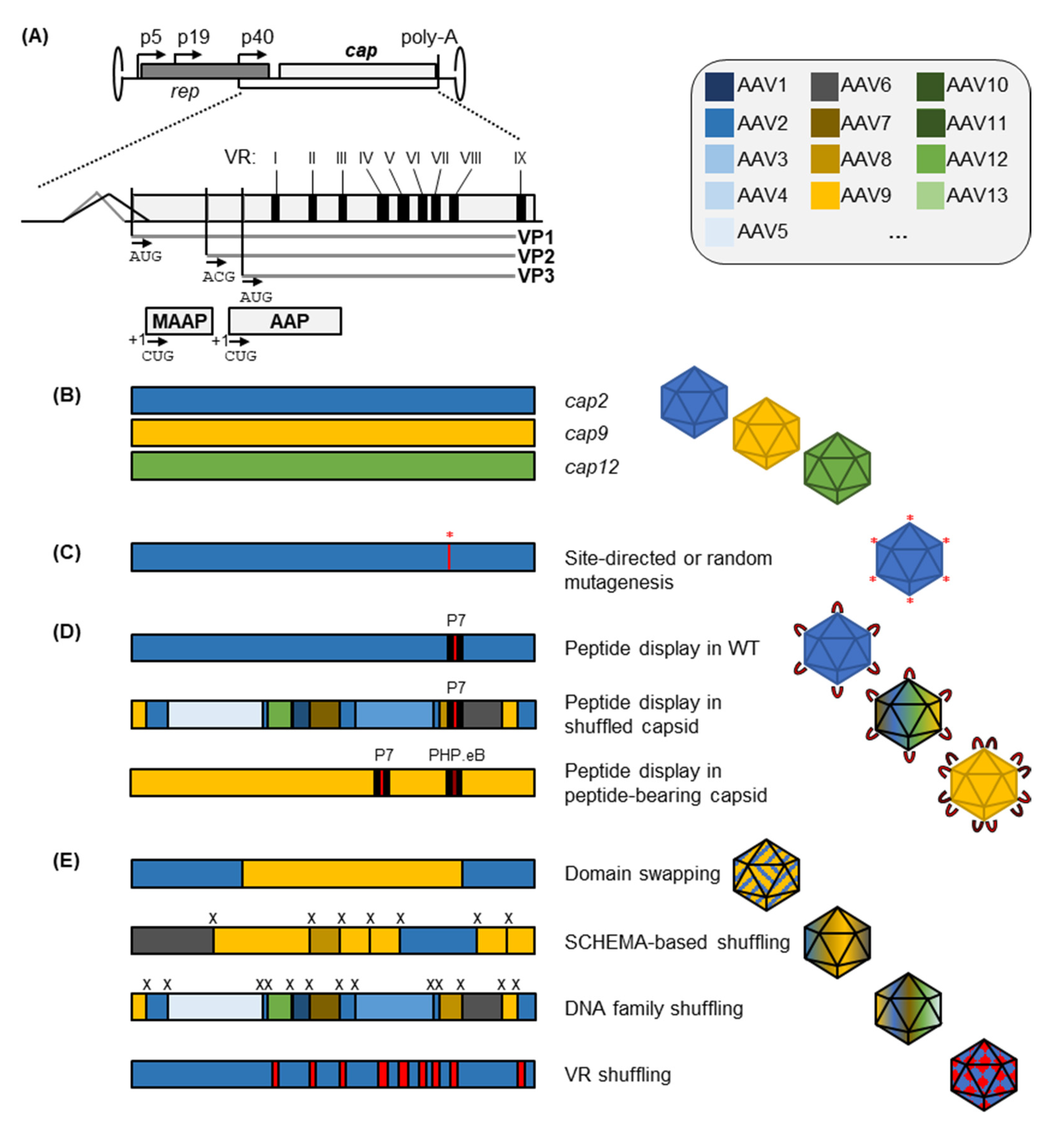
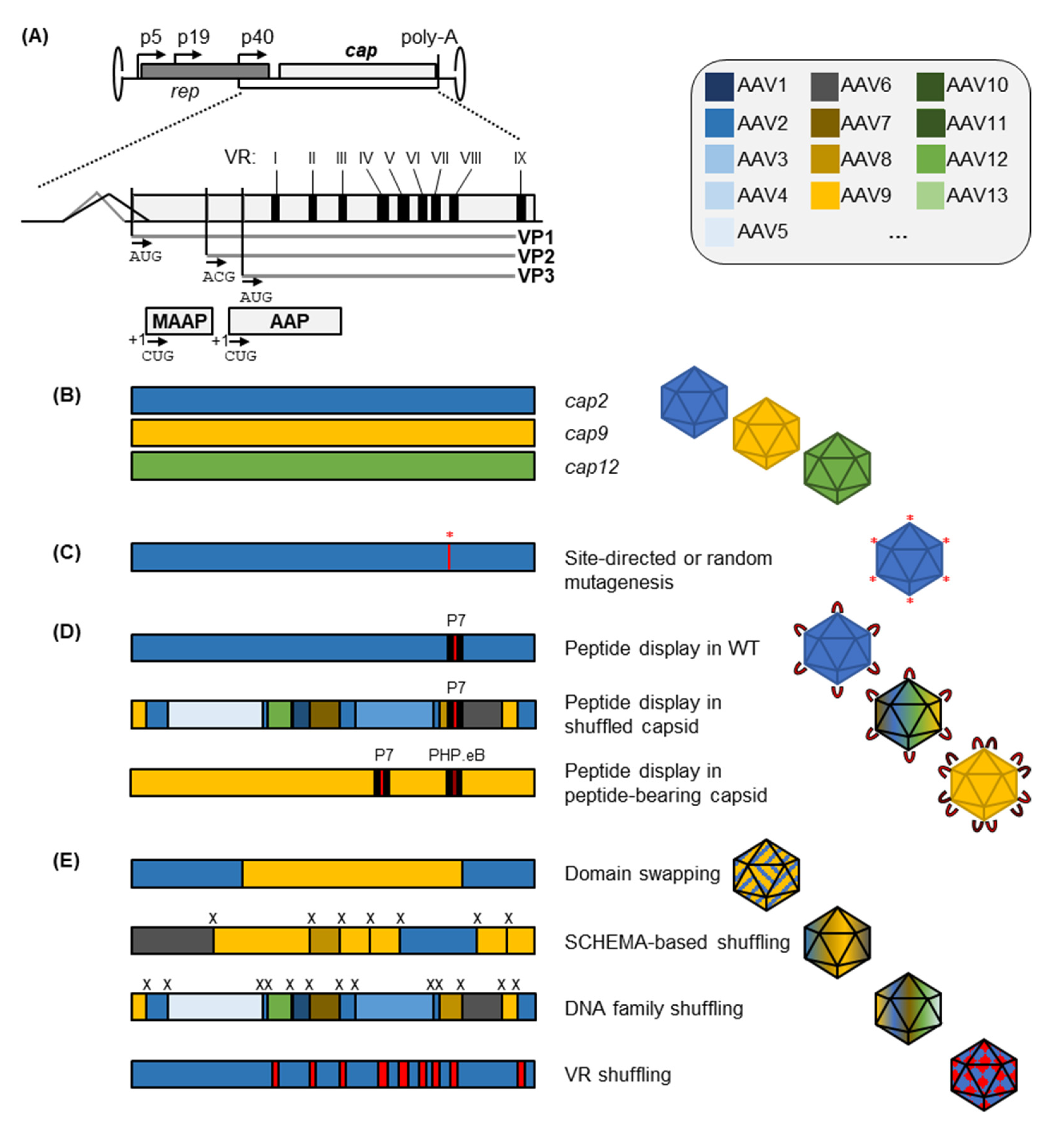
2. Capsid Engineering to Replace or Expand Natural AAV Serotypes
2.1. Peptide Insertion or Replacement for AAV Capsid Retargeting
2.1.1. Peptide Display in Serotypes Other than AAV2
2.1.2. Non-Random Peptide Screens
2.2. Integrating Features of Different Wild-Type Capsids into Engineered Progeny
2.2.1. Rational or Partially Randomized Integration of Residues from Multiple Serotypes through Domain and VR Swapping
2.2.2. DNA Family Shuffling for AAV Vector Evolution
2.2.3. Combinatorial Shuffling and Domain Swapping
3. New Synthetic Biology-Inspired Approaches
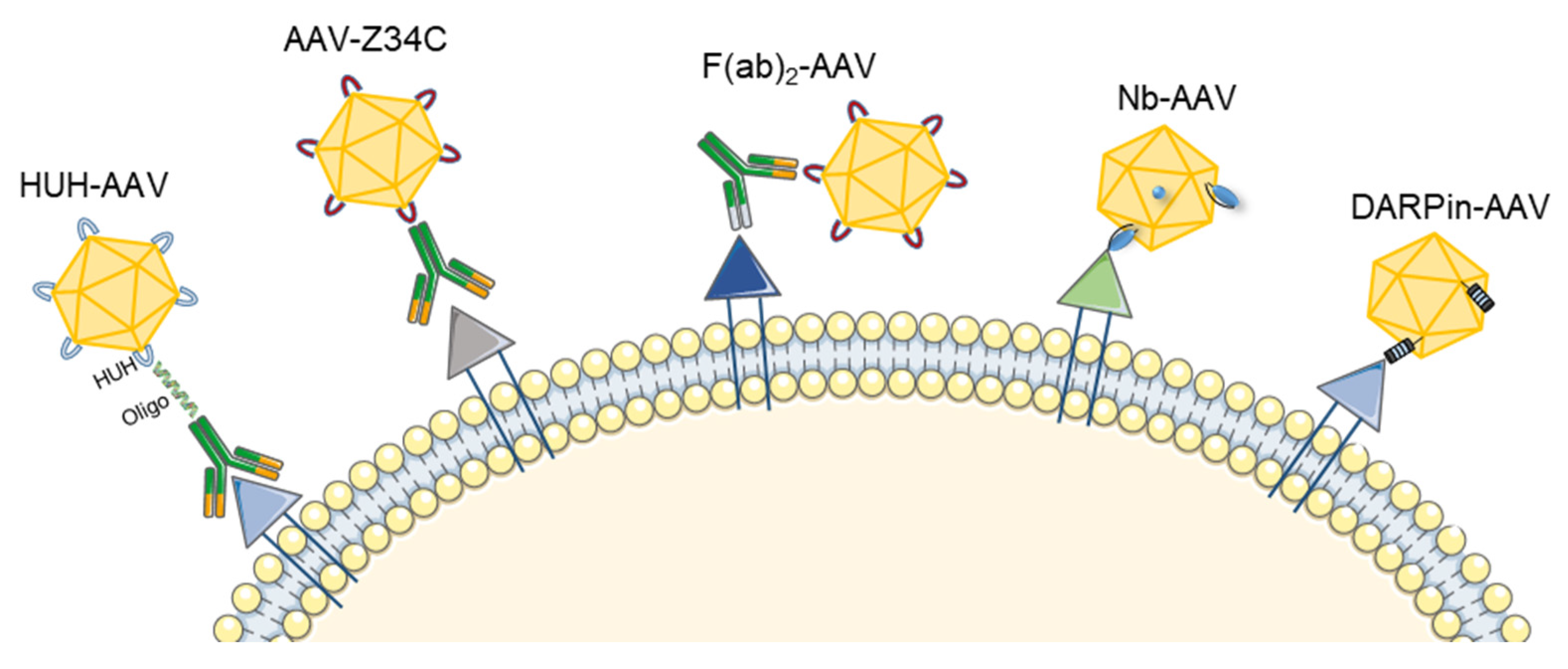
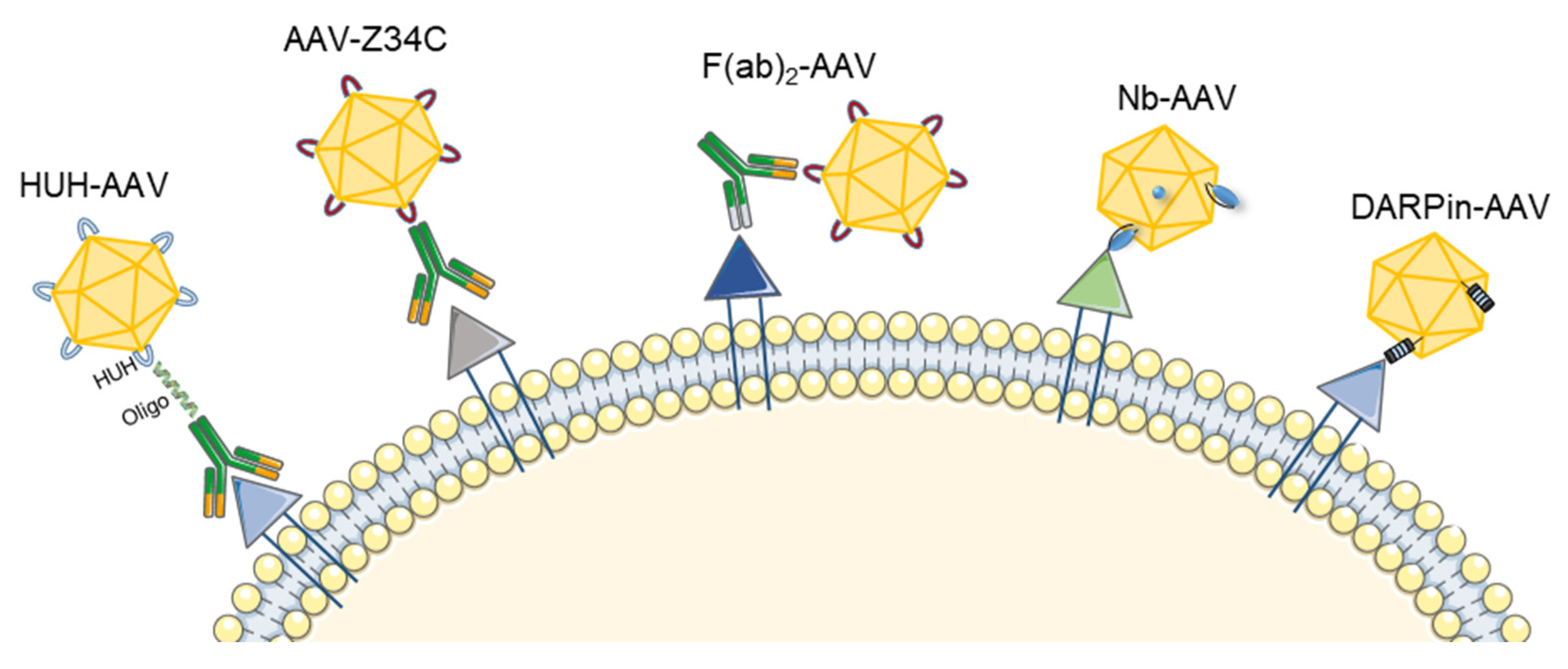
3.1. Antibody-Mediated AAV Retargeting
3.1.1. Genetic Fusion of Single-Chain Variable Fragments (scFv) and Ligands to the AAV Capsid
3.1.2. Use of Bispecific Antibodies as Bridging Molecules
3.1.3. Generation of Universal Templates Based on Antibody Binding Domains
3.1.4. Covalent Binding of Antibodies to the AAV Capsid Surface
3.2. DARPin-Mediated Viral Vector Retargeting
3.3. Nanobody-Mediated Targeting of AAV Gene Therapy Vectors
4. Data-Driven Capsid and Library Design
4.1. Machine Learning in AAV Library Generation
4.2. Ongoing Ventures in ML-Based Engineering of AAVs
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Marchini, A.; Bonifati, S.; Scott, E.M.; Angelova, A.L.; Rommelaere, J. Oncolytic parvoviruses: From basic virology to clinical applications. Virol. J. 2015, 12, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxwell, I.H.; Maxwell, F.; Rhode, S.L., III; Corsini, J.; Carlson, J.O. Recombinant LuIII autonomous parvovirus as a transient transducing vector for human cells. Hum. Gene Ther. 1993, 4, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Daya, S.; Berns, K.I. Gene therapy using adeno-associated virus vectors. Clin. Microbiol. Rev. 2008, 21, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Gene Therapy Clinical Trials Worldwide. Available online: https://a873679.fmphost.com/fmi/webd/GTCT (accessed on 4 May 2022).
- High-dose AAV gene therapy deaths. Nat. Biotechnol. 2020, 38, 910. [CrossRef]
- Au, H.K.E.; Isalan, M.; Mielcarek, M. Gene Therapy Advances: A Meta-Analysis of AAV Usage in Clinical Settings. Front. Med. 2022, 8, 809118. [Google Scholar] [CrossRef]
- Domenger, C.; Grimm, D. Next-generation AAV vectors—Do not judge a virus (only) by its cover. Hum. Mol. Genet. 2019, 28, R3–R14. [Google Scholar] [CrossRef] [Green Version]
- Powell, S.K.; Rivera-Soto, R.; Gray, S.J. Viral expression cassette elements to enhance transgene target specificity and expression in gene therapy. Discov. Med. 2015, 19, 49–57. [Google Scholar] [PubMed]
- Ozelo, M.C.; Mahlangu, J.; Pasi, K.J.; Giermasz, A.; Leavitt, A.D.; Laffan, M.; Symington, E.; Quon, D.V.; Wang, J.-D.; Peerlinck, K.; et al. Valoctocogene Roxaparvovec Gene Therapy for Hemophilia A. N. Engl. J. Med. 2022, 386, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.; Byrne, B.; Shieh, P.; Salabarria, S.; Berthy, J.; Corti, M.; Redican, S.; Lawrence, J.; Brown, K.; Shanks, C.; et al. Clinical trial highlights: O.2 ignite DMD Phase I/II ascending dose study of SGT-001 microdystrophin gene therapy for DMD: 1.5-year functional outcomes update. Neuromuscul. Disord. 2021, 31, S47. [Google Scholar] [CrossRef]
- Rapti, K.; Grimm, D. Adeno-Associated Viruses (AAV) and Host Immunity—A Race Between the Hare and the Hedgehog. Front. Immunol. 2021, 12, 753467. [Google Scholar] [CrossRef]
- Geisler, A.; Jungmann, A.; Kurreck, J.; Poller, W.; Katus, H.A.; Vetter, R.; Fechner, H.; Müller, O.J. microRNA122-regulated transgene expression increases specificity of cardiac gene transfer upon intravenous delivery of AAV9 vectors. Gene Ther. 2011, 18, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Ameres, S.L.; Friedline, R.; Hung, J.H.; Zhang, Y.; Xie, Q.; Zhong, L.; Su, Q.; He, R.; Li, M.; et al. Long-term, efficient inhibition of microRNA function in mice using rAAV vectors. Nat. Methods 2012, 9, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Dupaty, L.; Aillot, L.; Zhang, L.; Gallien, C.; Hallek, M.; Odenthal, M.; Adriouch, S.; Salvetti, A.; Büning, H. Vector uncoating limits adeno-associated viral vector-mediated transduction of human dendritic cells and vector immunogenicity. Sci. Rep. 2019, 9, 3631. [Google Scholar] [CrossRef] [PubMed]
- Gawel, D.R.; Serra-Musach, J.; Lilja, S.; Aagesen, J.; Arenas, A.; Asking, B.; Bengner, M.; Bjorkander, J.; Biggs, S.; Ernerudh, J.; et al. A validated single-cell-based strategy to identify diagnostic and therapeutic targets in complex diseases. Genome Med. 2019, 11, 47. [Google Scholar] [CrossRef]
- Luo, G.; Gao, Q.; Zhang, S.; Yan, B. Probing infectious disease by single-cell RNA sequencing: Progresses and perspectives. Comput. Struct. Biotechnol. J. 2020, 18, 2962–2971. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, Q.; He, J.; Li, Y. Novel Therapeutic Targets in Liver Fibrosis. Front. Mol. Biosci. 2021, 8, 766855. [Google Scholar] [CrossRef]
- Yang, S.; Zhou, J.; Li, D. Functions and Diseases of the Retinal Pigment Epithelium. Front. Pharmacol. 2021, 12, 727870. [Google Scholar] [CrossRef]
- Mamelak, M. Parkinson’s Disease, the Dopaminergic Neuron and Gammahydroxybutyrate. Neurol. Ther. 2018, 7, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Olson, E.N. Toward the correction of muscular dystrophy by gene editing. Proc. Natl. Acad. Sci. USA 2021, 118, e2004840117. [Google Scholar] [CrossRef]
- Plasschaert, L.W.; Zilionis, R.; Choo-Wing, R.; Savova, V.; Knehr, J.; Roma, G.; Klein, A.M.; Jaffe, A.B. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 2018, 560, 377–381. [Google Scholar] [CrossRef]
- Nonnenmacher, M.; Wang, W.; Child, M.A.; Ren, X.-Q.; Huang, C.; Ren, A.Z.; Tocci, J.; Chen, Q.; Bittner, K.; Tyson, K.; et al. Rapid evolution of blood-brain-barrier-penetrating AAV capsids by RNA-driven biopanning. Mol. Ther. Methods Clin. Dev. 2021, 20, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Tabebordbar, M.; Lagerborg, K.A.; Stanton, A.; King, E.M.; Ye, S.; Tellez, L.; Krunnfusz, A.; Tavakoli, S.; Widrick, J.J.; Messemer, K.A.; et al. Directed evolution of a family of AAV capsid variants enabling potent muscle-directed gene delivery across species. Cell 2021, 184, 4919–4938.e22. [Google Scholar] [CrossRef] [PubMed]
- Ravindra Kumar, S.; Miles, T.F.; Chen, X.; Brown, D.; Dobreva, T.; Huang, Q.; Ding, X.; Luo, Y.; Einarsson, P.H.; Greenbaum, A.; et al. Multiplexed Cre-dependent selection yields systemic AAVs for targeting distinct brain cell types. Nat. Methods 2020, 17, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Tabula Sapiens, C.; Jones, R.C.; Karkanias, J.; Krasnow, M.A.; Pisco, A.O.; Quake, S.R.; Salzman, J.; Yosef, N.; Bulthaup, B.; Brown, P.; et al. The Tabula Sapiens: A multiple-organ, single-cell transcriptomic atlas of humans. Science 2022, 376, eabl4896. [Google Scholar] [CrossRef]
- Ozturk, B.E.; Johnson, M.E.; Kleyman, M.; Turunc, S.; He, J.; Jabalameli, S.; Xi, Z.; Visel, M.; Dufour, V.L.; Iwabe, S.; et al. scAAVengr, a transcriptome-based pipeline for quantitative ranking of engineered AAVs with single-cell resolution. Elife 2021, 10, e64175. [Google Scholar] [CrossRef]
- Kremer, L.P.M.; Cerrizuela, S.; Dehler, S.; Stiehl, T.; Weinmann, J.; Abendroth, H.; Kleber, S.; Laure, A.; El Andari, J.; Anders, S.; et al. High throughput screening of novel AAV capsids identifies variants for transduction of adult NSCs within the subventricular zone. Mol. Ther. Methods Clin. Dev. 2021, 23, 33–50. [Google Scholar] [CrossRef]
- Brown, D.; Altermatt, M.; Dobreva, T.; Chen, S.; Wang, A.; Thomson, M.; Gradinaru, V. Deep Parallel Characterization of AAV Tropism and AAV-Mediated Transcriptional Changes via Single-Cell RNA Sequencing. Front. Immunol. 2021, 12, 730825. [Google Scholar] [CrossRef]
- Eichhoff, A.M.; Borner, K.; Albrecht, B.; Schafer, W.; Baum, N.; Haag, F.; Korbelin, J.; Trepel, M.; Braren, I.; Grimm, D.; et al. Nanobody-Enhanced Targeting of AAV Gene Therapy Vectors. Mol. Ther. Methods Clin. Dev. 2019, 15, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Michels, A.; Frank, A.M.; Gunther, D.M.; Mataei, M.; Borner, K.; Grimm, D.; Hartmann, J.; Buchholz, C.J. Lentiviral and adeno-associated vectors efficiently transduce mouse T lymphocytes when targeted to murine CD8. Mol. Ther. Methods Clin. Dev. 2021, 23, 334–347. [Google Scholar] [CrossRef]
- Jackson, C.B.; Richard, A.S.; Ojha, A.; Conkright, K.A.; Trimarchi, J.M.; Bailey, C.C.; Alpert, M.D.; Kay, M.A.; Farzan, M.; Choe, H. AAV vectors engineered to target insulin receptor greatly enhance intramuscular gene delivery. Mol. Ther. Methods Clin. Dev. 2020, 19, 496–506. [Google Scholar] [CrossRef]
- Zdechlik, A.C.; He, Y.; Aird, E.J.; Gordon, W.R.; Schmidt, D. Programmable Assembly of Adeno-Associated Virus-Antibody Composites for Receptor-Mediated Gene Delivery. Bioconjugate Chem. 2020, 31, 1093–1106. [Google Scholar] [CrossRef]
- Keeler, A.M.; Flotte, T.R. Recombinant Adeno-Associated Virus Gene Therapy in Light of Luxturna (and Zolgensma and Glybera): Where Are We, and How Did We Get Here? Annu. Rev. Virol. 2019, 6, 601–621. [Google Scholar] [CrossRef]
- Kuzmin, D.A.; Shutova, M.V.; Johnston, N.R.; Smith, O.P.; Fedorin, V.V.; Kukushkin, Y.S.; van der Loo, J.C.M.; Johnstone, E.C. The clinical landscape for AAV gene therapies. Nat. Rev. Drug Discov. 2021, 20, 173–174. [Google Scholar] [CrossRef] [PubMed]
- Louis Jeune, V.; Joergensen, J.A.; Hajjar, R.J.; Weber, T. Pre-existing Anti–Adeno-Associated Virus Antibodies as a Challenge in AAV Gene Therapy. Hum. Gene Ther. Methods 2013, 24, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Govindasamy, L.; Padron, E.; McKenna, R.; Muzyczka, N.; Kaludov, N.; Chiorini John, A.; Agbandje-McKenna, M. Structurally Mapping the Diverse Phenotype of Adeno-Associated Virus Serotype 4. J. Virol. 2006, 80, 11556–11570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogden, P.J.; Kelsic, E.D.; Sinai, S.; Church, G.M. Comprehensive AAV capsid fitness landscape reveals a viral gene and enables machine-guided design. Science 2019, 366, 1139–1143. [Google Scholar] [CrossRef] [PubMed]
- Sonntag, F.; Schmidt, K.; Kleinschmidt Jürgen, A. A viral assembly factor promotes AAV2 capsid formation in the nucleolus. Proc. Natl. Acad. Sci. USA 2010, 107, 10220–10225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kern, A.; Schmidt, K.; Leder, C.; Müller, O.J.; Wobus, C.E.; Bettinger, K.; Von der Lieth, C.W.; King, J.A.; Kleinschmidt, J.A. Identification of a Heparin-Binding Motif on Adeno-Associated Virus Type 2 Capsids. J. Virol. 2003, 77, 11072–11081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, O.J.; Kaul, F.; Weitzman, M.D.; Pasqualini, R.; Arap, W.; Kleinschmidt, J.A.; Trepel, M. Random peptide libraries displayed on adeno-associated virus to select for targeted gene therapy vectors. Nat. Biotechnol. 2003, 21, 1040. [Google Scholar] [CrossRef]
- Tan, F.; Chu, C.; Qi, J.; Li, W.; You, D.; Li, K.; Chen, X.; Zhao, W.; Cheng, C.; Liu, X.; et al. AAV-ie enables safe and efficient gene transfer to inner ear cells. Nat. Commun. 2019, 10, 3733. [Google Scholar] [CrossRef] [Green Version]
- Goertsen, D.; Flytzanis, N.C.; Goeden, N.; Chuapoco, M.R.; Cummins, A.; Chen, Y.; Fan, Y.; Zhang, Q.; Sharma, J.; Duan, Y.; et al. AAV capsid variants with brain-wide transgene expression and decreased liver targeting after intravenous delivery in mouse and marmoset. Nat. Neurosci. 2022, 25, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Storm, T.; Kay, M.A. Characterization of the Relationship of AAV Capsid Domain Swapping to Liver Transduction Efficiency. Mol. Ther. 2007, 15, 1955–1962. [Google Scholar] [CrossRef] [PubMed]
- Ojala, D.S.; Sun, S.; Santiago-Ortiz, J.L.; Shapiro, M.G.; Romero, P.A.; Schaffer, D.V. In Vivo Selection of a Computationally Designed SCHEMA AAV Library Yields a Novel Variant for Infection of Adult Neural Stem Cells in the SVZ. Mol. Ther. 2018, 26, 304–319. [Google Scholar] [CrossRef] [Green Version]
- Grimm, D.; Lee, J.S.; Wang, L.; Desai, T.; Akache, B.; Storm, T.A.; Kay, M.A. In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J. Virol. 2008, 82, 5887–5911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsic, D.; Govindasamy, L.; Currlin, S.; Markusic, D.M.; Tseng, Y.-S.; Herzog, R.W.; Agbandje-McKenna, M.; Zolotukhin, S. Vector Design Tour de Force: Integrating Combinatorial and Rational Approaches to Derive Novel Adeno-associated Virus Variants. Mol. Ther. 2014, 22, 1900–1909. [Google Scholar] [CrossRef] [Green Version]
- Korneyenkov, M.A.; Zamyatnin, A.A., Jr. Next Step in Gene Delivery: Modern Approaches and Further Perspectives of AAV Tropism Modification. Pharmaceutics 2021, 13, 750. [Google Scholar] [CrossRef]
- Zincarelli, C.; Soltys, S.; Rengo, G.; Rabinowitz, J.E. Analysis of AAV Serotypes 1–9 Mediated Gene Expression and Tropism in Mice After Systemic Injection. Mol. Ther. 2008, 16, 1073–1080. [Google Scholar] [CrossRef]
- Börner, K.; Kienle, E.; Huang, L.-Y.; Weinmann, J.; Sacher, A.; Bayer, P.; Stüllein, C.; Fakhiri, J.; Zimmermann, L.; Westhaus, A.; et al. Pre-arrayed Pan-AAV Peptide Display Libraries for Rapid Single-Round Screening. Mol. Ther. 2020, 28, 1016–1032. [Google Scholar] [CrossRef]
- Landegger, L.D.; Pan, B.; Askew, C.; Wassmer, S.J.; Gluck, S.D.; Galvin, A.; Taylor, R.; Forge, A.; Stankovic, K.M.; Holt, J.R.; et al. A synthetic AAV vector enables safe and efficient gene transfer to the mammalian inner ear. Nat. Biotechnol. 2017, 35, 280–284. [Google Scholar] [CrossRef] [Green Version]
- Grimm, D.; Büning, H. Small But Increasingly Mighty: Latest Advances in AAV Vector Research, Design, and Evolution. Hum. Gene Ther. 2017, 28, 1075–1086. [Google Scholar] [CrossRef]
- Zolotukhin, S.; Vandenberghe, L.H. AAV capsid design: A Goldilocks challenge. Trends Mol. Med. 2022, 28, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.; Zolotukhin, S. E Pluribus Unum: 50 Years of Research, Millions of Viruses, and One Goal—Tailored Acceleration of AAV Evolution. Mol. Ther. 2015, 23, 1819–1831. [Google Scholar] [CrossRef] [Green Version]
- Hudry, E.; Vandenberghe, L.H. Therapeutic AAV Gene Transfer to the Nervous System: A Clinical Reality. Neuron 2019, 101, 839–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol. Ther. Methods Clin. Dev. 2018, 8, 87–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, C.; Scheideler, O.; Schaffer, D. Engineering the AAV capsid to evade immune responses. Curr. Opin. Biotechnol. 2019, 60, 99–103. [Google Scholar] [CrossRef]
- Li, C.; Samulski, R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020, 21, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef]
- Samulski, R.J.; Berns, K.I.; Tan, M.; Muzyczka, N. Cloning of adeno-associated virus into pBR322: Rescue of intact virus from the recombinant plasmid in human cells. Proc. Natl. Acad. Sci. USA 1982, 79, 2077–2081. [Google Scholar] [CrossRef] [Green Version]
- Girod, A.; Ried, M.; Wobus, C.; Lahm, H.; Leike, K.; Kleinschmidt, J.; Deléage, G.; Hallek, M. Genetic capsid modifications allow efficient re-targeting of adeno-associated virus type 2. Nat. Med. 1999, 5, 1052–1056. [Google Scholar] [CrossRef]
- Rabinowitz, J.E.; Xiao, W.; Samulski, R.J. Insertional Mutagenesis of AAV2 Capsid and the Production of Recombinant Virus. Virology 1999, 265, 274–285. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Xiao, W.; Conlon, T.; Hughes, J.; Agbandje-McKenna, M.; Ferkol, T.; Flotte, T.; Muzyczka, N. Mutational Analysis of the Adeno-Associated Virus Type 2 (AAV2) Capsid Gene and Construction of AAV2 Vectors with Altered Tropism. J. Virol. 2000, 74, 8635–8647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, W.; Arnold, G.S.; Bartlett, J.S. Insertional Mutagenesis of the Adeno-Associated Virus Type 2 (AAV2) Capsid Gene and Generation of AAV2 Vectors Targeted to Alternative Cell-Surface Receptors. Hum. Gene Ther. 2001, 12, 1697–1711. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Bu, W.; Bhatia, S.; Hare, J.; Somasundaram, T.; Azzi, A.; Chapman Michael, S. The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy. Proc. Natl. Acad. Sci. USA 2002, 99, 10405–10410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opie, S.R.; Warrington, K.H., Jr.; Agbandje-McKenna, M.; Zolotukhin, S.; Muzyczka, N. Identification of amino acid residues in the capsid proteins of adeno-associated virus type 2 that contribute to heparan sulfate proteoglycan binding. J. Virol. 2003, 77, 6995–7006. [Google Scholar] [CrossRef] [Green Version]
- Perabo, L.; Goldnau, D.; White, K.; Endell, J.; Boucas, J.; Humme, S.; Work Lorraine, M.; Janicki, H.; Hallek, M.; Baker Andrew, H.; et al. Heparan Sulfate Proteoglycan Binding Properties of Adeno-Associated Virus Retargeting Mutants and Consequences for Their In Vivo Tropism. J. Virol. 2006, 80, 7265–7269. [Google Scholar] [CrossRef] [Green Version]
- Boucas, J.; Lux, K.; Huber, A.; Schievenbusch, S.; von Freyend, M.J.; Perabo, L.; Quadt-Humme, S.; Odenthal, M.; Hallek, M.; Büning, H. Engineering adeno-associated virus serotype 2-based targeting vectors using a new insertion site-position 453-and single point mutations. J. Gene Med. 2009, 11, 1103–1113. [Google Scholar] [CrossRef]
- Adachi, K.; Enoki, T.; Kawano, Y.; Veraz, M.; Nakai, H. Drawing a high-resolution functional map of adeno-associated virus capsid by massively parallel sequencing. Nat. Commun. 2014, 5, 3075. [Google Scholar] [CrossRef]
- Büning, H.; Srivastava, A. Capsid Modifications for Targeting and Improving the Efficacy of AAV Vectors. Mol. Ther. Methods Clin. Dev. 2019, 12, 248–265. [Google Scholar] [CrossRef]
- Perabo, L.; Büning, H.; Kofler, D.M.; Ried, M.U.; Girod, A.; Wendtner, C.M.; Enssle, J.; Hallek, M. In vitro selection of viral vectors with modified tropism: The adeno-associated virus display. Mol. Ther. 2003, 8, 151–157. [Google Scholar] [CrossRef]
- Körbelin, J.; Sieber, T.; Michelfelder, S.; Lunding, L.; Spies, E.; Hunger, A.; Alawi, M.; Rapti, K.; Indenbirken, D.; Müller, O.J.; et al. Pulmonary Targeting of Adeno-associated Viral Vectors by Next-generation Sequencing-guided Screening of Random Capsid Displayed Peptide Libraries. Mol. Ther. 2016, 24, 1050–1061. [Google Scholar] [CrossRef] [Green Version]
- Dalkara, D.; Byrne, L.C.; Klimczak, R.R.; Visel, M.; Yin, L.; Merigan, W.H.; Flannery, J.G.; Schaffer, D.V. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci. Transl. Med. 2013, 5, 189ra76. [Google Scholar] [CrossRef] [PubMed]
- Khabou, H.; Desrosiers, M.; Winckler, C.; Fouquet, S.; Auregan, G.; Bemelmans, A.-P.; Sahel, J.-A.; Dalkara, D. Insight into the mechanisms of enhanced retinal transduction by the engineered AAV2 capsid variant -7m8. Biotechnol. Bioeng. 2016, 113, 2712–2724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murlidharan, G.; Samulski, R.J.; Asokan, A. Biology of adeno-associated viral vectors in the central nervous system. Front. Mol. Neurosci. 2014, 7, 76. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Kauss, M.A.; Kopin, E.; Chandra, M.; Ul-Hasan, T.; Miller, E.; Jayandharan, G.R.; Rivers, A.E.; Aslanidi, G.V.; Ling, C.; et al. Optimizing the transduction efficiency of capsid-modified AAV6 serotype vectors in primary human hematopoietic stem cells in vitro and in a xenograft mouse model in vivo. Cytotherapy 2013, 15, 986–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinmann, J.; Weis, S.; Sippel, J.; Tulalamba, W.; Remes, A.; El Andari, J.; Herrmann, A.-K.; Pham, Q.H.; Borowski, C.; Hille, S.; et al. Identification of a myotropic AAV by massively parallel in vivo evaluation of barcoded capsid variants. Nat. Commun. 2020, 11, 5432. [Google Scholar] [CrossRef] [PubMed]
- Kotchey, N.M.; Adachi, K.; Zahid, M.; Inagaki, K.; Charan, R.; Parker, R.S.; Nakai, H. A Potential Role of Distinctively Delayed Blood Clearance of Recombinant Adeno-associated Virus Serotype 9 in Robust Cardiac Transduction. Mol. Ther. 2011, 19, 1079–1089. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Li, S.; Wang, H.; Guo, Y.; Gessler, D.J.; Cao, C.; Su, Q.; Kramer, J.; Zhong, L.; Ahmed, S.S.; et al. Global CNS transduction of adult mice by intravenously delivered rAAVrh.8 and rAAVrh.10 and nonhuman primates by rAAVrh.10. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 1299–1309. [Google Scholar] [CrossRef] [Green Version]
- Merkel, S.F.; Andrews, A.M.; Lutton, E.M.; Mu, D.; Hudry, E.; Hyman, B.T.; Maguire, C.A.; Ramirez, S.H. Trafficking of adeno-associated virus vectors across a model of the blood-brain barrier; a comparative study of transcytosis and transduction using primary human brain endothelial cells. J. Neurochem. 2017, 140, 216–230. [Google Scholar] [CrossRef] [Green Version]
- Deverman, B.E.; Pravdo, P.L.; Simpson, B.P.; Kumar, S.R.; Chan, K.Y.; Banerjee, A.; Wu, W.-L.; Yang, B.; Huber, N.; Pasca, S.P.; et al. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 2016, 34, 204–209. [Google Scholar] [CrossRef]
- Chan, K.Y.; Jang, M.J.; Yoo, B.B.; Greenbaum, A.; Ravi, N.; Wu, W.-L.; Sánchez-Guardado, L.; Lois, C.; Mazmanian, S.K.; Deverman, B.E.; et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 2017, 20, 1172–1179. [Google Scholar] [CrossRef]
- Hordeaux, J.; Yuan, Y.; Clark, P.M.; Wang, Q.; Martino, R.A.; Sims, J.J.; Bell, P.; Raymond, A.; Stanford, W.L.; Wilson, J.M. The GPI-Linked Protein LY6A Drives AAV-PHP.B Transport across the Blood-Brain Barrier. Mol. Ther. 2019, 27, 912–921. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Chan, K.Y.; Tobey, I.G.; Chan, Y.A.; Poterba, T.; Boutros, C.L.; Balazs, A.B.; Daneman, R.; Bloom, J.M.; Seed, C.; et al. Delivering genes across the blood-brain barrier: LY6A, a novel cellular receptor for AAV-PHP.B capsids. PLoS ONE 2019, 14, e0225206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzaki, Y.; Konno, A.; Mochizuki, R.; Shinohara, Y.; Nitta, K.; Okada, Y.; Hirai, H. Intravenous administration of the adeno-associated virus-PHP.B capsid fails to upregulate transduction efficiency in the marmoset brain. Neurosci. Lett. 2018, 665, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Batista, A.R.; King, O.D.; Reardon, C.P.; Davis, C.; Shankaracharya, F.; Philip, V.; Gray-Edwards, H.; Aronin, N.; Lutz, C.; Landers, J.; et al. Ly6a Differential Expression in Blood-Brain Barrier Is Responsible for Strain Specific Central Nervous System Transduction Profile of AAV-PHP.B. Hum. Gene Ther. 2020, 31, 90–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hordeaux, J.; Wang, Q.; Katz, N.; Buza, E.L.; Bell, P.; Wilson, J.M. The Neurotropic Properties of AAV-PHP.B Are Limited to C57BL/6J Mice. Mol. Ther. 2018, 26, 664–668. [Google Scholar] [CrossRef] [PubMed]
- Loughner, C.L.; Bruford, E.A.; McAndrews, M.S.; Delp, E.E.; Swamynathan, S.; Swamynathan, S.K. Organization, evolution and functions of the human and mouse Ly6/uPAR family genes. Hum. Genom. 2016, 10, 10. [Google Scholar] [CrossRef] [Green Version]
- Hanlon, K.S.; Meltzer, J.C.; Buzhdygan, T.; Cheng, M.J.; Sena-Esteves, M.; Bennett, R.E.; Sullivan, T.P.; Razmpour, R.; Gong, Y.; Ng, C.; et al. Selection of an Efficient AAV Vector for Robust CNS Transgene Expression. Mol. Ther. Methods Clin. Dev. 2019, 15, 320–332. [Google Scholar] [CrossRef] [Green Version]
- Westhaus, A.; Cabanes-Creus, M.; Jonker, T.; Sallard, E.; Navarro, R.G.; Zhu, E.; Baltazar Torres, G.; Lee, S.; Wilmott, P.; Gonzalez-Cordero, A.; et al. AAV-p40 Bioengineering Platform for Variant Selection Based on Transgene Expression. Hum. Gene Ther. 2022, 33, 664–682. [Google Scholar] [CrossRef]
- Martino, R.A.; Fluck Edwin, C.; Murphy, J.; Wang, Q.; Hoff, H.; Pumroy Ruth, A.; Lee Claudia, Y.; Sims Joshua, J.; Roy, S.; Moiseenkova-Bell Vera, Y.; et al. Context-Specific Function of the Engineered Peptide Domain of PHP.B. J. Virol. 2021, 95, e01164-21. [Google Scholar] [CrossRef]
- Tao, Y.; Liu, X.; Yang, L.; Chu, C.; Tan, F.; Yu, Z.; Ke, J.; Li, X.; Zheng, X.; Zhao, X.; et al. AAV-ie-K558R mediated cochlear gene therapy and hair cell regeneration. Signal Transduct. Target Ther. 2022, 7, 109. [Google Scholar] [CrossRef]
- Davidsson, M.; Wang, G.; Aldrin-Kirk, P.; Cardoso, T.; Nolbrant, S.; Hartnor, M.; Mudannayake, J.; Parmar, M.; Björklund, T. A systematic capsid evolution approach performed in vivo for the design of AAV vectors with tailored properties and tropism. Proc. Natl. Acad. Sci. USA 2019, 116, 27053–27062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tse Longping, V.; Klinc Kelli, A.; Madigan Victoria, J.; Castellanos Rivera Ruth, M.; Wells Lindsey, F.; Havlik, L.P.; Smith, J.K.; Agbandje-McKenna, M.; Asokan, A. Structure-guided evolution of antigenically distinct adeno-associated virus variants for immune evasion. Proc. Natl. Acad. Sci. USA 2017, 114, E4812–E4821. [Google Scholar] [CrossRef] [Green Version]
- Havlik, L.P.; Simon, K.E.; Smith, J.K.; Klinc, K.A.; Tse, L.V.; Oh, D.K.; Fanous, M.M.; Meganck, R.M.; Mietzsch, M.; Kleinschmidt, J.; et al. Coevolution of Adeno-associated Virus Capsid Antigenicity and Tropism through a Structure-Guided Approach. J. Virol. 2020, 94, e00976-20. [Google Scholar] [CrossRef] [PubMed]
- Emmanuel Shanan, N.; Smith, J.K.; Hsi, J.; Tseng, Y.-S.; Kaplan, M.; Mietzsch, M.; Chipman, P.; Asokan, A.; McKenna, R.; Agbandje-McKenna, M.; et al. Structurally Mapping Antigenic Epitopes of Adeno-associated Virus 9: Development of Antibody Escape Variants. J. Virol. 2022, 96, e01251-21. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Alvira Mauricio, R.; Somanathan, S.; Lu, Y.; Vandenberghe Luk, H.; Rux John, J.; Calcedo, R.; Sanmiguel, J.; Abbas, Z.; Wilson James, M. Adeno-associated viruses undergo substantial evolution in primates during natural infections. Proc. Natl. Acad. Sci. USA 2003, 100, 6081–6086. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.; Vandenberghe, H.L.; Wilson, M.J. New Recombinant Serotypes of AAV Vectors. Curr. Gene Ther. 2005, 5, 285–297. [Google Scholar] [CrossRef]
- Gao, G.; Vandenberghe, L.H.; Alvira, M.R.; Lu, Y.; Calcedo, R.; Zhou, X.; Wilson, J.M. Clades of Adeno-associated viruses are widely disseminated in human tissues. J. Virol. 2004, 78, 6381–6388. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.-P.; Alvira, M.R.; Wang, L.; Calcedo, R.; Johnston, J.; Wilson, J.M. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. USA 2002, 99, 11854–11859. [Google Scholar] [CrossRef] [Green Version]
- Vandenberghe, L.H.; Breous, E.; Nam, H.J.; Gao, G.; Xiao, R.; Sandhu, A.; Johnston, J.; Debyser, Z.; Agbandje-McKenna, M.; Wilson, J.M. Naturally occurring singleton residues in AAV capsid impact vector performance and illustrate structural constraints. Gene Ther. 2009, 16, 1416–1428. [Google Scholar] [CrossRef] [Green Version]
- Fakhiri, J.; Linse, K.P.; Mietzsch, M.; Xu, M.; Schneider, M.A.; Meister, M.; Schildgen, O.; Schnitzler, P.; Soderlund-Venermo, M.; Agbandje-McKenna, M.; et al. Impact of Natural or Synthetic Singletons in the Capsid of Human Bocavirus 1 on Particle Infectivity and Immunoreactivity. J. Virol. 2020, 94, e00170-20. [Google Scholar] [CrossRef]
- Castellanos, M.; Perez, R.; Carrasco, C.; Hernando-Perez, M.; Gomez-Herrero, J.; de Pablo, P.J.; Mateu, M.G. Mechanical elasticity as a physical signature of conformational dynamics in a virus particle. Proc. Natl. Acad. Sci. USA 2012, 109, 12028–12033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinn, E.; Pacouret, S.; Khaychuk, V.; Turunen, H.T.; Carvalho, L.S.; Andres-Mateos, E.; Shah, S.; Shelke, R.; Maurer, A.C.; Plovie, E.; et al. In Silico Reconstruction of the Viral Evolutionary Lineage Yields a Potent Gene Therapy Vector. Cell Rep. 2015, 12, 1056–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santiago-Ortiz, J.; Ojala, D.S.; Westesson, O.; Weinstein, J.R.; Wong, S.Y.; Steinsapir, A.; Kumar, S.; Holmes, I.; Schaffer, D.V. AAV ancestral reconstruction library enables selection of broadly infectious viral variants. Gene Ther. 2015, 22, 934–946. [Google Scholar] [CrossRef] [Green Version]
- Pan, B.; Askew, C.; Galvin, A.; Heman-Ackah, S.; Asai, Y.; Indzhykulian, A.A.; Jodelka, F.M.; Hastings, M.L.; Lentz, J.J.; Vandenberghe, L.H.; et al. Gene therapy restores auditory and vestibular function in a mouse model of Usher syndrome type 1c. Nat. Biotechnol. 2017, 35, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Hashimoto, K.; Xiao, R.; Vandenberghe, L.H.; Liberman, M.C. Cochlear gene therapy with ancestral AAV in adult mice: Complete transduction of inner hair cells without cochlear dysfunction. Sci. Rep. 2017, 7, 45524. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Xiao, R.; Andres-Mateos, E.; Vandenberghe, L.H. Single stranded adeno-associated virus achieves efficient gene transfer to anterior segment in the mouse eye. PLoS ONE 2017, 12, e0182473. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, L.S.; Xiao, R.; Wassmer, S.J.; Langsdorf, A.; Zinn, E.; Pacouret, S.; Shah, S.; Comander, J.I.; Kim, L.A.; Lim, L.; et al. Synthetic Adeno-Associated Viral Vector Efficiently Targets Mouse and Nonhuman Primate Retina In Vivo. Hum. Gene Ther. 2018, 29, 771–784. [Google Scholar] [CrossRef]
- Hauck, B.; Xiao, W. Characterization of Tissue Tropism Determinants of Adeno-Associated Virus Type 1. J. Virol. 2003, 77, 2768–2774. [Google Scholar] [CrossRef] [Green Version]
- Biswas, M.; Marsic, D.; Li, N.; Zou, C.; Gonzalez-Aseguinolaza, G.; Zolotukhin, I.; Kumar, S.R.P.; Rana, J.; Butterfield, J.S.S.; Kondratov, O.; et al. Engineering and In Vitro Selection of a Novel AAV3B Variant with High Hepatocyte Tropism and Reduced Seroreactivity. Mol. Ther. Methods Clin. Dev. 2020, 19, 347–361. [Google Scholar] [CrossRef]
- Voigt, C.A.; Martinez, C.; Wang, Z.-G.; Mayo, S.L.; Arnold, F.H. Protein building blocks preserved by recombination. Nat. Struct. Biol. 2002, 9, 553–558. [Google Scholar] [CrossRef]
- Ho, M.L.; Adler, B.A.; Torre, M.L.; Silberg, J.J.; Suh, J. SCHEMA Computational Design of Virus Capsid Chimeras: Calibrating How Genome Packaging, Protection, and Transduction Correlate with Calculated Structural Disruption. ACS Synth. Biol. 2013, 2, 724–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabanes-Creus, M.; Ginn, S.L.; Amaya, A.K.; Liao, S.H.Y.; Westhaus, A.; Hallwirth, C.V.; Wilmott, P.; Ward, J.; Dilworth, K.L.; Santilli, G.; et al. Codon-Optimization of Wild-Type Adeno-Associated Virus Capsid Sequences Enhances DNA Family Shuffling while Conserving Functionality. Mol. Ther. Methods Clin. Dev. 2019, 12, 71–84. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Asokan, A.; Wu, Z.; Van Dyke, T.; DiPrimio, N.; Johnson, J.S.; Govindaswamy, L.; Agbandje-McKenna, M.; Leichtle, S.; Redmond, D.E., Jr.; et al. Engineering and selection of shuffled AAV genomes: A new strategy for producing targeted biological nanoparticles. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- Koerber, J.T.; Jang, J.H.; Schaffer, D.V. DNA shuffling of adeno-associated virus yields functionally diverse viral progeny. Mol. Ther. 2008, 16, 1703–1709. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, A.-K.; Bender, C.; Kienle, E.; Grosse, S.; El Andari, J.; Botta, J.; Schürmann, N.; Wiedtke, E.; Niopek, D.; Grimm, D. A Robust and All-Inclusive Pipeline for Shuffling of Adeno-Associated Viruses. ACS Synth. Biol. 2019, 8, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Maguire, C.A.; Gianni, D.; Meijer, D.H.; Shaket, L.A.; Wakimoto, H.; Rabkin, S.D.; Gao, G.; Sena-Esteves, M. Directed evolution of adeno-associated virus for glioma cell transduction. J. Neuro Oncol. 2010, 96, 337–347. [Google Scholar] [CrossRef]
- Yang, L.; Jiang, J.; Drouin Lauren, M.; Agbandje-Mckenna, M.; Chen, C.; Qiao, C.; Pu, D.; Hu, X.; Wang, D.-Z.; Li, J.; et al. A myocardium tropic adeno-associated virus (AAV) evolved by DNA shuffling and in vivo selection. Proc. Natl. Acad. Sci. USA 2009, 106, 3946–3951. [Google Scholar] [CrossRef] [Green Version]
- Gray, S.J.; Blake, B.L.; Criswell, H.E.; Nicolson, S.C.; Samulski, R.J.; McCown, T.J. Directed Evolution of a Novel Adeno-associated Virus (AAV) Vector That Crosses the Seizure-compromised Blood–Brain Barrier (BBB). Mol. Ther. 2010, 18, 570–578. [Google Scholar] [CrossRef]
- Lisowski, L.; Dane, A.P.; Chu, K.; Zhang, Y.; Cunningham, S.C.; Wilson, E.M.; Nygaard, S.; Grompe, M.; Alexander, I.E.; Kay, M.A. Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature 2014, 506, 382–386. [Google Scholar] [CrossRef] [Green Version]
- Baruteau, J.; Cunningham, S.C.; Yilmaz, B.S.; Perocheau, D.P.; Eaglestone, S.; Burke, D.; Thrasher, A.J.; Waddington, S.N.; Lisowski, L.; Alexander, I.E.; et al. Safety and efficacy of an engineered hepatotropic AAV gene therapy for ornithine transcarbamylase deficiency in cynomolgus monkeys. Mol. Ther. Methods Clin. Dev. 2021, 23, 135–146. [Google Scholar] [CrossRef]
- Paulk, N.K.; Pekrun, K.; Zhu, E.; Nygaard, S.; Li, B.; Xu, J.; Chu, K.; Leborgne, C.; Dane, A.P.; Haft, A.; et al. Bioengineered AAV Capsids with Combined High Human Liver Transduction In Vivo and Unique Humoral Seroreactivity. Mol. Ther. 2018, 26, 289–303. [Google Scholar] [CrossRef] [Green Version]
- Pekrun, K.; De Alencastro, G.; Luo, Q.-J.; Liu, J.; Kim, Y.; Nygaard, S.; Galivo, F.; Zhang, F.; Song, R.; Tiffany, M.R.; et al. Using a barcoded AAV capsid library to select for clinically relevant gene therapy vectors. JCI Insight 2019, 4, e131610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Alencastro, G.; Pekrun, K.; Valdmanis, P.; Tiffany, M.; Xu, J.; Kay, M.A. Tracking Adeno-Associated Virus Capsid Evolution by High-Throughput Sequencing. Hum. Gene Ther. 2020, 31, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Cabanes-Creus, M.; Navarro, R.G.; Liao, S.H.Y.; Baltazar, G.; Drouyer, M.; Zhu, E.; Scott, S.; Luong, C.; Wilson, L.O.W.; Alexander, I.E.; et al. Single amino acid insertion allows functional transduction of murine hepatocytes with human liver tropic AAV capsids. Mol. Ther. Methods Clin. Dev. 2021, 21, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Cabanes-Creus, M.; Navarro, R.G.; Zhu, E.; Baltazar, G.; Liao, S.H.Y.; Drouyer, M.; Amaya, A.K.; Scott, S.; Nguyen, L.H.; Westhaus, A.; et al. Novel human liver-tropic AAV variants define transferable domains that markedly enhance the human tropism of AAV7 and AAV8. Mol. Ther. Methods Clin. Dev. 2022, 24, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Albright, B.H.; Storey, C.M.; Murlidharan, G.; Castellanos Rivera, R.M.; Berry, G.E.; Madigan, V.J.; Asokan, A. Mapping the Structural Determinants Required for AAVrh.10 Transport across the Blood-Brain Barrier. Mol. Ther. 2018, 26, 510–523. [Google Scholar] [CrossRef] [Green Version]
- Katrekar, D.; Moreno, A.M.; Chen, G.; Worlikar, A.; Mali, P. Oligonucleotide conjugated multi-functional adeno-associated viruses. Sci. Rep. 2018, 8, 3589. [Google Scholar] [CrossRef]
- Horowitz, E.D.; Weinberg, M.S.; Asokan, A. Glycated AAV vectors: Chemical redirection of viral tissue tropism. Bioconjugate Chem. 2011, 22, 529–532. [Google Scholar] [CrossRef] [Green Version]
- Pearce, H.A.; Qian, H.; Connell, T.U.; Huang, D.; Gottstein, C.; Donnelly, P.S.; Peter, K.; Gregorevic, P.; Hagemeyer, C.E. Site-Specific Glycation and Chemo-enzymatic Antibody Sortagging for the Retargeting of rAAV6 to Inflamed Endothelium. Mol. Ther. Methods Clin. Dev. 2019, 14, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Mevel, M.; Bouzelha, M.; Leray, A.; Pacouret, S.; Guilbaud, M.; Penaud-Budloo, M.; Alvarez-Dorta, D.; Dubreil, L.; Gouin, S.G.; Combal, J.P.; et al. Chemical modification of the adeno-associated virus capsid to improve gene delivery. Chem. Sci. 2019, 11, 1122–1131. [Google Scholar] [CrossRef] [Green Version]
- Reul, J.; Muik, A.; Buchholz, C.J. Ligand Coupling to the AAV Capsid for Cell-Specific Gene Transfer. Methods Mol. Biol. 2019, 1950, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.K.; Maheshri, N.; Kaspar, B.; Schaffer, D.V. PEG conjugation moderately protects adeno-associated viral vectors against antibody neutralization. Biotechnol. Bioeng. 2005, 92, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Zhou, X.; Zhang, C.; Yu, X.; Tian, Z.; Zhang, L.; Zhou, D. Site-Specific PEGylated Adeno-Associated Viruses with Increased Serum Stability and Reduced Immunogenicity. Molecules 2017, 22, 1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ried, M.U.; Girod, A.; Leike, K.; Buning, H.; Hallek, M. Adeno-associated virus capsids displaying immunoglobulin-binding domains permit antibody-mediated vector retargeting to specific cell surface receptors. J. Virol. 2002, 76, 4559–4566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartlett, J.S.; Kleinschmidt, J.; Boucher, R.C.; Samulski, R.J. Targeted adeno-associated virus vector transduction of nonpermissive cells mediated by a bispecific F(ab’gamma)2 antibody. Nat. Biotechnol. 1999, 17, 181–186. [Google Scholar] [CrossRef]
- Kuklik, J.; Michelfelder, S.; Schiele, F.; Kreuz, S.; Lamla, T.; Muller, P.; Park, J.E. Development of a Bispecific Antibody-Based Platform for Retargeting of Capsid Modified AAV Vectors. Int. J. Mol. Sci. 2021, 22, 8355. [Google Scholar] [CrossRef]
- Munch, R.C.; Janicki, H.; Volker, I.; Rasbach, A.; Hallek, M.; Buning, H.; Buchholz, C.J. Displaying high-affinity ligands on adeno-associated viral vectors enables tumor cell-specific and safe gene transfer. Mol. Ther. 2013, 21, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Munch, R.C.; Muth, A.; Muik, A.; Friedel, T.; Schmatz, J.; Dreier, B.; Trkola, A.; Pluckthun, A.; Buning, H.; Buchholz, C.J. Off-target-free gene delivery by affinity-purified receptor-targeted viral vectors. Nat. Commun. 2015, 6, 6246. [Google Scholar] [CrossRef] [Green Version]
- Muik, A.; Reul, J.; Friedel, T.; Muth, A.; Hartmann, K.P.; Schneider, I.C.; Munch, R.C.; Buchholz, C.J. Covalent coupling of high-affinity ligands to the surface of viral vector particles by protein trans-splicing mediates cell type-specific gene transfer. Biomaterials 2017, 144, 84–94. [Google Scholar] [CrossRef]
- Castelli, M.S.; McGonigle, P.; Hornby, P.J. The pharmacology and therapeutic applications of monoclonal antibodies. Pharmacol. Res. Perspect. 2019, 7, e00535. [Google Scholar] [CrossRef]
- Yang, Q.; Mamounas, M.; Yu, G.; Kennedy, S.; Leaker, B.; Merson, J.; Wong-Staal, F.; Yu, M.; Barber, J.R. Development of novel cell surface CD34-targeted recombinant adenoassociated virus vectors for gene therapy. Hum. Gene Ther. 1998, 9, 1929–1937. [Google Scholar] [CrossRef] [PubMed]
- Grifman, M.; Trepel, M.; Speece, P.; Gilbert, L.B.; Arap, W.; Pasqualini, R.; Weitzman, M.D. Incorporation of tumor-targeting peptides into recombinant adeno-associated virus capsids. Mol. Ther. 2001, 3, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Warrington, K.H., Jr.; Gorbatyuk, O.S.; Harrison, J.K.; Opie, S.R.; Zolotukhin, S.; Muzyczka, N. Adeno-associated virus type 2 VP2 capsid protein is nonessential and can tolerate large peptide insertions at its N terminus. J. Virol. 2004, 78, 6595–6609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lux, K.; Goerlitz, N.; Schlemminger, S.; Perabo, L.; Goldnau, D.; Endell, J.; Leike, K.; Kofler, D.M.; Finke, S.; Hallek, M.; et al. Green fluorescent protein-tagged adeno-associated virus particles allow the study of cytosolic and nuclear trafficking. J. Virol. 2005, 79, 11776–11787. [Google Scholar] [CrossRef] [Green Version]
- Asokan, A.; Johnson, J.S.; Li, C.; Samulski, R.J. Bioluminescent virion shells: New tools for quantitation of AAV vector dynamics in cells and live animals. Gene Ther. 2008, 15, 1618–1622. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Chen, K.; Lei, Q.; Ma, P.; Yuan, A.Q.; Zhao, Y.; Jiang, Y.; Fang, H.; Xing, S.; Fang, Y.; et al. The state of the art of bispecific antibodies for treating human malignancies. EMBO Mol. Med. 2021, 13, e14291. [Google Scholar] [CrossRef]
- Wobus, C.E.; Hugle-Dorr, B.; Girod, A.; Petersen, G.; Hallek, M.; Kleinschmidt, J.A. Monoclonal antibodies against the adeno-associated virus type 2 (AAV-2) capsid: Epitope mapping and identification of capsid domains involved in AAV-2-cell interaction and neutralization of AAV-2 infection. J. Virol. 2000, 74, 9281–9293. [Google Scholar] [CrossRef] [Green Version]
- Gigout, L.; Rebollo, P.; Clement, N.; Warrington, K.H.; Muzyczka, N.; Linden, R.M.; Weber, T. Altering AAV tropism with mosaic viral capsids. Mol. Ther. 2005, 11, 856–865. [Google Scholar] [CrossRef]
- Diamandis, E.P.; Christopoulos, T.K. The biotin-(strept)avidin system: Principles and applications in biotechnology. Clin. Chem. 1991, 37, 625–636. [Google Scholar] [CrossRef]
- Stachler, M.D.; Chen, I.; Ting, A.Y.; Bartlett, J.S. Site-specific modification of AAV vector particles with biophysical probes and targeting ligands using biotin ligase. Mol. Ther. 2008, 16, 1467–1473. [Google Scholar] [CrossRef]
- Arnold, G.S.; Sasser, A.K.; Stachler, M.D.; Bartlett, J.S. Metabolic biotinylation provides a unique platform for the purification and targeting of multiple AAV vector serotypes. Mol. Ther. 2006, 14, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Dundas, C.M.; Demonte, D.; Park, S. Streptavidin-biotin technology: Improvements and innovations in chemical and biological applications. Appl. Microbiol. Biotechnol. 2013, 97, 9343–9353. [Google Scholar] [CrossRef] [PubMed]
- Schechter, B.; Silberman, R.; Arnon, R.; Wilchek, M. Tissue distribution of avidin and streptavidin injected to mice. Effect of avidin carbohydrate, streptavidin truncation and exogenous biotin. Eur. J. Biochem. 1990, 189, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.K.; Frabutt, D.; Li, L.; Xiao, W. Chemical Modifications of the Capsid for Redirecting and Improving the Efficacy of Adeno-Associated Virus Vectors. Hum. Gene Ther. 2021, 32, 1433–1438. [Google Scholar] [CrossRef]
- Wei, F.; McConnell, K.I.; Yu, T.K.; Suh, J. Conjugation of paclitaxel on adeno-associated virus (AAV) nanoparticles for co-delivery of genes and drugs. Eur. J. Pharm. Sci. 2012, 46, 167–172. [Google Scholar] [CrossRef]
- Liu, Y.; Fang, Y.; Zhou, Y.; Zandi, E.; Lee, C.L.; Joo, K.I.; Wang, P. Site-specific modification of adeno-associated viruses via a genetically engineered aldehyde tag. Small 2013, 9, 421–429. [Google Scholar] [CrossRef]
- Lovendahl, K.N.; Hayward, A.N.; Gordon, W.R. Sequence-Directed Covalent Protein-DNA Linkages in a Single Step Using HUH-Tags. J. Am. Chem. Soc. 2017, 139, 7030–7035. [Google Scholar] [CrossRef] [Green Version]
- Sedgwick, S.G.; Smerdon, S.J. The ankyrin repeat: A diversity of interactions on a common structural framework. Trends Biochem. Sci. 1999, 24, 311–316. [Google Scholar] [CrossRef]
- Schweizer, A.; Rusert, P.; Berlinger, L.; Ruprecht, C.R.; Mann, A.; Corthesy, S.; Turville, S.G.; Aravantinou, M.; Fischer, M.; Robbiani, M.; et al. CD4-specific designed ankyrin repeat proteins are novel potent HIV entry inhibitors with unique characteristics. PLoS Pathog. 2008, 4, e1000109. [Google Scholar] [CrossRef] [Green Version]
- Stefan, N.; Martin-Killias, P.; Wyss-Stoeckle, S.; Honegger, A.; Zangemeister-Wittke, U.; Pluckthun, A. DARPins recognizing the tumor-associated antigen EpCAM selected by phage and ribosome display and engineered for multivalency. J. Mol. Biol. 2011, 413, 826–843. [Google Scholar] [CrossRef]
- Binz, H.K.; Amstutz, P.; Kohl, A.; Stumpp, M.T.; Briand, C.; Forrer, P.; Grütter, M.G.; Plückthun, A. High-affinity binders selected from designed ankyrin repeat protein libraries. Nat. Biotechnol. 2004, 22, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Munch, R.C.; Muhlebach, M.D.; Schaser, T.; Kneissl, S.; Jost, C.; Pluckthun, A.; Cichutek, K.; Buchholz, C.J. DARPins: An efficient targeting domain for lentiviral vectors. Mol. Ther. 2011, 19, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Reul, J.; Frisch, J.; Engeland, C.E.; Thalheimer, F.B.; Hartmann, J.; Ungerechts, G.; Buchholz, C.J. Tumor-Specific Delivery of Immune Checkpoint Inhibitors by Engineered AAV Vectors. Front. Oncol. 2019, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, J.; Thalheimer, F.B.; Hopfner, F.; Kerzel, T.; Khodosevich, K.; Garcia-Gonzalez, D.; Monyer, H.; Diester, I.; Buning, H.; Carette, J.E.; et al. GluA4-Targeted AAV Vectors Deliver Genes Selectively to Interneurons while Relying on the AAV Receptor for Entry. Mol. Ther. Methods Clin. Dev. 2019, 14, 252–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinderer, C.; Katz, N.; Buza, E.L.; Dyer, C.; Goode, T.; Bell, P.; Richman, L.K.; Wilson, J.M. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum. Gene Ther. 2018, 29, 285–298. [Google Scholar] [CrossRef] [Green Version]
- Morales, L.; Gambhir, Y.; Bennett, J.; Stedman, H.H. Broader Implications of Progressive Liver Dysfunction and Lethal Sepsis in Two Boys following Systemic High-Dose AAV. Mol. Ther. 2020, 28, 1753–1755. [Google Scholar] [CrossRef]
- Wilson, J.M.; Flotte, T.R. Moving Forward After Two Deaths in a Gene Therapy Trial of Myotubular Myopathy. Hum. Gene Ther. 2020, 31, 695–696. [Google Scholar] [CrossRef] [PubMed]
- Stone, D.; Kenkel, E.J.; Loprieno, M.A.; Tanaka, M.; De Silva Feelixge, H.S.; Kumar, A.J.; Stensland, L.; Obenza, W.M.; Wangari, S.; Ahrens, C.Y.; et al. Gene Transfer in Adeno-Associated Virus Seropositive Rhesus Macaques Following Rapamycin Treatment and Subcutaneous Delivery of AAV6, but Not Retargeted AAV6 Vectors. Hum. Gene Ther. 2021, 32, 96–112. [Google Scholar] [CrossRef] [PubMed]
- Angelova, A.L.; Geletneky, K.; Nuesch, J.P.; Rommelaere, J. Tumor Selectivity of Oncolytic Parvoviruses: From in vitro and Animal Models to Cancer Patients. Front. Bioeng. Biotechnol. 2015, 3, 55. [Google Scholar] [CrossRef] [Green Version]
- Bretscher, C.; Marchini, A. H-1 Parvovirus as a Cancer-Killing Agent: Past, Present, and Future. Viruses 2019, 11, 562. [Google Scholar] [CrossRef] [Green Version]
- Fakhiri, J.; Grimm, D. Best of most possible worlds: Hybrid gene therapy vectors based on parvoviruses and heterologous viruses. Mol. Ther. 2021, 29, 3359–3382. [Google Scholar] [CrossRef] [PubMed]
- Bannas, P.; Hambach, J.; Koch-Nolte, F. Nanobodies and Nanobody-Based Human Heavy Chain Antibodies As Antitumor Therapeutics. Front. Immunol. 2017, 8, 1603. [Google Scholar] [CrossRef] [PubMed]
- Perruchini, C.; Pecorari, F.; Bourgeois, J.P.; Duyckaerts, C.; Rougeon, F.; Lafaye, P. Llama VHH antibody fragments against GFAP: Better diffusion in fixed tissues than classical monoclonal antibodies. Acta Neuropathol. 2009, 118, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Judd, J.; Wei, F.; Nguyen, P.Q.; Tartaglia, L.J.; Agbandje-McKenna, M.; Silberg, J.J.; Suh, J. Random Insertion of mCherry Into VP3 Domain of Adeno-associated Virus Yields Fluorescent Capsids With no Loss of Infectivity. Mol. Ther. Nucleic Acids 2012, 1, e54. [Google Scholar] [CrossRef] [Green Version]
- Hamann, M.V.; Beschorner, N.; Vu, X.K.; Hauber, I.; Lange, U.C.; Traenkle, B.; Kaiser, P.D.; Foth, D.; Schneider, C.; Buning, H.; et al. Improved targeting of human CD4+ T cells by nanobody-modified AAV2 gene therapy vectors. PLoS ONE 2021, 16, e0261269. [Google Scholar] [CrossRef]
- Zhong, L.; Li, B.; Mah, C.S.; Govindasamy, L.; Agbandje-McKenna, M.; Cooper, M.; Herzog, R.W.; Zolotukhin, I.; Warrington, K.H., Jr.; Weigel-Van Aken, K.A.; et al. Next generation of adeno-associated virus 2 vectors: Point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc. Natl. Acad. Sci. USA 2008, 105, 7827–7832. [Google Scholar] [CrossRef] [Green Version]
- Toueille, M. Development of purification steps for several AAV serotypes using POROS™ CaptureSelect™ AAVX affinity chromatography. Cell Gene Ther. Insights 2018, 4, 637–645. [Google Scholar] [CrossRef]
- Nass, S.A.; Mattingly, M.A.; Woodcock, D.A.; Burnham, B.L.; Ardinger, J.A.; Osmond, S.E.; Frederick, A.M.; Scaria, A.; Cheng, S.H.; O’Riordan, C.R. Universal Method for the Purification of Recombinant AAV Vectors of Differing Serotypes. Mol. Ther Methods Clin. Dev. 2018, 9, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Lock, M.; Prongay, A.J.; Alvira, M.R.; Petkov, B.; Wilson, J.M. Identification of an adeno-associated virus binding epitope for AVB sepharose affinity resin. Mol. Ther. Methods Clin. Dev. 2015, 2, 15040. [Google Scholar] [CrossRef]
- Greener, J.G.; Kandathil, S.M.; Moffat, L.; Jones, D.T. A guide to machine learning for biologists. Nat. Rev. Mol. Cell Biol. 2022, 23, 40–55. [Google Scholar] [CrossRef]
- Libbrecht, M.W.; Noble, W.S. Machine learning applications in genetics and genomics. Nat. Rev. Genet. 2015, 16, 321–332. [Google Scholar] [CrossRef] [Green Version]
- Webb, S. Deep learning for biology. Nature 2018, 554, 555–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.K.; Wu, Z.; Arnold, F.H. Machine-learning-guided directed evolution for protein engineering. Nat. Methods 2019, 16, 687–694. [Google Scholar] [CrossRef]
- Bryant, D.H.; Bashir, A.; Sinai, S.; Jain, N.K.; Ogden, P.J.; Riley, P.F.; Church, G.M.; Colwell, L.J.; Kelsic, E.D. Deep diversification of an AAV capsid protein by machine learning. Nat. Biotechnol. 2021, 39, 691–696. [Google Scholar] [CrossRef]
- Marques, A.D.; Kummer, M.; Kondratov, O.; Banerjee, A.; Moskalenko, O.; Zolotukhin, S. Applying machine learning to predict viral assembly for adeno-associated virus capsid libraries. Mol. Ther. Methods Clin. Dev. 2021, 20, 276–286. [Google Scholar] [CrossRef]
- Sinai, S.; Jain, N.; Church, G.M.; Kelsic, E.D. Generative AAV capsid diversification by latent interpolation. bioRxiv 2021. [Google Scholar] [CrossRef]
- Mikos, G.; Chen, W.; Suh, J. Machine Learning Identification of Capsid Mutations to Improve AAV Production Fitness. bioRxiv 2021. [Google Scholar] [CrossRef]
- Zhu, D.; Brookes, D.H.; Busia, A.; Carneiro, A.; Fannjiang, C.; Popova, G.; Shin, D.; Chang, E.F.; Nowakowski, T.J.; Listgarten, J.; et al. Machine learning-based library design improves packaging and diversity of adeno-associated virus (AAV) libraries. bioRxiv 2021. [Google Scholar] [CrossRef]
- Kelsic, E.D.; Church, G.M. Challenges and opportunities of machine-guided capsid engineering for gene therapy. Cell Gene Ther. Insights 2019, 5, 523–536. [Google Scholar] [CrossRef]
- Wec, A.Z.; Lin, K.S.; Kwasnieski, J.C.; Sinai, S.; Gerold, J.; Kelsic, E.D. Overcoming Immunological Challenges Limiting Capsid-Mediated Gene Therapy With Machine Learning. Front. Immunol. 2021, 12, 674021. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.; Slavov, N. Single cell protein analysis for systems biology. Essays Biochem. 2018, 62, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Westhaus, A.; Cabanes-Creus, M.; Rybicki, A.; Baltazar, G.; Navarro, R.G.; Zhu, E.; Drouyer, M.; Knight, M.; Albu, R.F.; Ng, B.H.; et al. High-Throughput In Vitro, Ex Vivo, and In Vivo Screen of Adeno-Associated Virus Vectors Based on Physical and Functional Transduction. Hum. Gene Ther. 2020, 31, 575–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becker, J.; Fakhiri, J.; Grimm, D. Fantastic AAV Gene Therapy Vectors and How to Find Them—Random Diversification, Rational Design and Machine Learning. Pathogens 2022, 11, 756. https://doi.org/10.3390/pathogens11070756
Becker J, Fakhiri J, Grimm D. Fantastic AAV Gene Therapy Vectors and How to Find Them—Random Diversification, Rational Design and Machine Learning. Pathogens. 2022; 11(7):756. https://doi.org/10.3390/pathogens11070756
Chicago/Turabian StyleBecker, Jonas, Julia Fakhiri, and Dirk Grimm. 2022. "Fantastic AAV Gene Therapy Vectors and How to Find Them—Random Diversification, Rational Design and Machine Learning" Pathogens 11, no. 7: 756. https://doi.org/10.3390/pathogens11070756
APA StyleBecker, J., Fakhiri, J., & Grimm, D. (2022). Fantastic AAV Gene Therapy Vectors and How to Find Them—Random Diversification, Rational Design and Machine Learning. Pathogens, 11(7), 756. https://doi.org/10.3390/pathogens11070756