
Paramyxovirus Diversity within One Population of Miniopterus fuliginosus Bats in Sri Lanka

 ,
,  , , , and
, , , and
Abstract
:1. Introduction
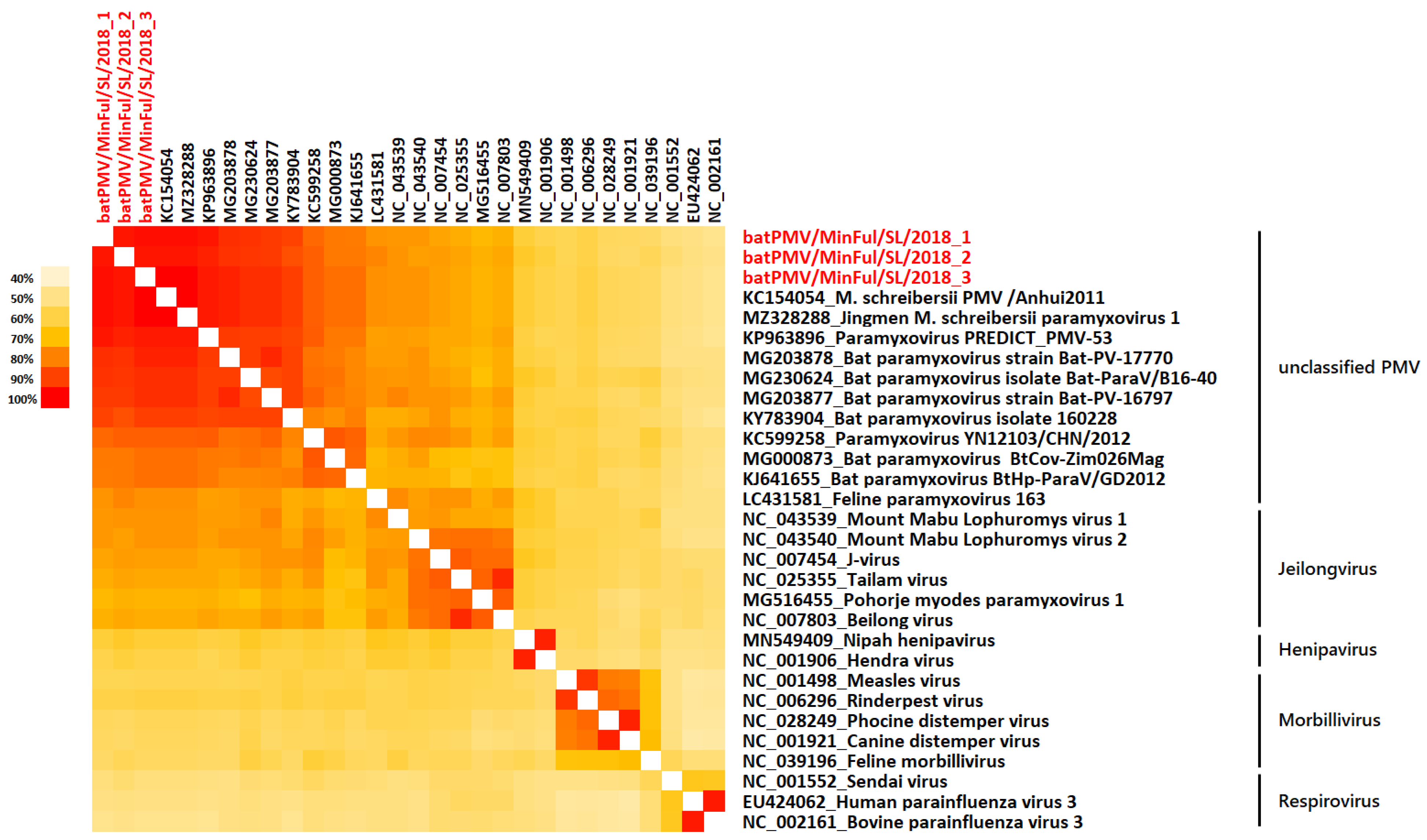
2. Results
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rima, B.; Balkema-Buschmann, A.; Dundon, W.G.; Duprex, P.; Easton, A.; Fouchier, R.; Kurath, G.; Lamb, R.; Lee, B.; Rota, P.; et al. ICTV Virus Taxonomy Profile: Paramyxoviridae. J. Gen. Virol. 2019, 100, 1593–1594. [Google Scholar] [CrossRef] [PubMed]
- Larsen, B.B.; Gryseels, S.; Otto, H.W.; Worobey, M. Evolution and Diversity of Bat and Rodent Paramyxoviruses from North America. J. Virol. 2021, 96, JVI-01098. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Corman, V.M.; Müller, M.A.; Maganga, G.D.; Vallo, P.; Binger, T.; Gloza-Rausch, F.; Rasche, A.; Yordanov, S.; Seebens, A.; et al. Bats Host Major Mammalian Paramyxoviruses. Nat. Commun. 2012, 3, 796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V. Learning from Bats to Escape from Potent or Severe Viral Infections. In SARS-CoV-2 Origin and COVID-19 Pandemic across the Globe; IntechOpen: London, UK, 2021. [Google Scholar] [CrossRef]
- Liang, J.; Zhu, C.; Zhang, L. Cospeciation of Coronavirus and Paramyxovirus with Their Bat Hosts in the Same Geographical Areas. BMC Ecol. Evo. 2021, 21, 148. [Google Scholar] [CrossRef]
- Roes, F.L. On the Evolution of Virulent Zoonotic Viruses in Bats. Biol. Theory 2020, 15, 223–225. [Google Scholar] [CrossRef]
- Samal, S.K. Paramyxoviruses of Animals. Encycl. Virol. 2008, 3, 40–47. [Google Scholar] [CrossRef]
- Yapa, W.B. A Field Guide to the Bats of Sri Lanka; Dilmah Ceylon Tea Company PLC: Colombo, Sri Lanka, 2017. [Google Scholar]
- Muzeniek, T.; Perera, T.; Siriwardana, S.; Bas, D.; Kaplan, F.; Öruc, M.; Becker-Ziaja, B.; Schwarz, F.; Premawansa, G.; Premawansa, S.; et al. Detection of Alpha-and Betacoronaviruses in Miniopterus Fuliginosus and Rousettus Leschenaultii, Two Species of Sri Lankan Bats. Vaccines 2021, 9, 650. [Google Scholar] [CrossRef]
- Kudagammana, H.D.W.S.; Thevanesam, V.; Chu, D.K.W.; Eriyagama, N.B.; Peiris, J.S.M.; Noordeen, F. Coronaviruses in Guano from Pteropus Medius Bats in Peradeniya, Sri Lanka. Transbound. Emerg. Dis. 2018, 65, 1122–1124. [Google Scholar] [CrossRef] [Green Version]
- Gunawardena, P.S.; Marston, D.A.; Ellis, R.J.; Wise, E.L.; Karawita, A.C.; Breed, A.C.; McElhinney, L.M.; Johnson, N.; Banyard, A.C.; Fooks, A.R. Lyssavirus in Indian Flying Foxes, Sri Lanka. Emerg. Infect. Dis. 2016, 22, 1456–1459. [Google Scholar] [CrossRef]
- Muzeniek, T.; Perera, T.; Siriwardana, S.; Bas, D.; Kaplan, F.; Öruc, M.; Becker-Ziaja, B.; Perera, I.; Weerasena, J.; Handunnetti, S.; et al. Full Genome of BatCoV/MinFul/2018/SriLanka, a Novel Alpha-Coronavirus Detected in Miniopterus Fuliginosus, Sri Lanka. Viruses 2022, 14, 337. [Google Scholar] [CrossRef]
- Kurth, A.; Kohl, C.; Brinkmann, A.; Ebinger, A.; Harper, J.A.; Wang, L.F.; Mühldorfer, K.; Wibbelt, G. Novel Paramyxoviruses in Free-Ranging European Bats. PLoS ONE 2012, 7, e38688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thibault, P.A.; Watkinson, R.E.; Moreira-Soto, A.; Drexler, J.F.; Lee, B. Zoonotic Potential of Emerging Paramyxoviruses: Knowns and Unknowns. Adv. Virus Res. 2017, 98, 1–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsh, G.A.; Wang, L.F. Hendra and Nipah Viruses: Why Are They so Deadly? Curr. Opin. Virol. 2012, 2, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, J.H.; Adkins, S.; Alioto, D.; Alkhovsky, S.V.; Amarasinghe, G.K.; Anthony, S.J.; Avšič-Županc, T.; Ayllón, M.A.; Bahl, J.; Balkema-Buschmann, A.; et al. 2020 taxonomic update for phylum Negarnaviricota (Riboviria: Orthornavirae), including the large orders Bunyavirales and Mononegavirales. Arch. Virol. 2020, 165, 3023–3027. [Google Scholar] [CrossRef]
- Amarasinghe, G.K.; Ayllón, M.A.; Bào, Y.; Basler, C.F.; Bavari, S.; Blasdell, K.R.; Briese, T.; Brown, P.A.; Bukreyev, A.; Balkema-Buschmann, A.; et al. Taxonomy of the Order Mononegavirales: Update 2019. Arch. Virol. 2019, 164, 1967–1980. [Google Scholar] [CrossRef] [Green Version]
- Rima, B.; Collins, P.; Easton, A.; Fouchier, R.; Kurath, G.; Lamb, R.A.; Lee, B.; Maisner, A.; Rota, P.; Wang, L.F. Problems of classification in the Family Paramyxoviridae. Arch. Virol. 2018, 163, 1395–1404. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.S.; Noh, J.Y.; Lo, V.T.; Choi, Y.G.; Yoon, S.W.; Jeong, D.G.; Kim, H.K. The Epidemiological Characteristics of the Korean Bat Paramyxovirus between 2016 and 2019. Microorganisms 2020, 8, 844. [Google Scholar] [CrossRef]
- Noh, J.Y.; Jeong, D.G.; Yoon, S.W.; Kim, J.H.; Choi, Y.G.; Kang, S.Y.; Kim, H.K. Isolation and characterization of novel bat paramyxovirus B16-40 potentially belonging to the proposed genus Shaanvirus. Sci. Rep. 2018, 8, 12533. [Google Scholar] [CrossRef]
- Peel, A.J.; Wells, K.; Giles, J.; Boyd, V.; Burroughs, A.; Edson, D.; Crameri, G.; Baker, M.L.; Field, H.; Wang, L.F.; et al. Synchronous Shedding of Multiple Bat Paramyxoviruses Coincides with Peak Periods of Hendra Virus Spillover. Emerg. Microbes Infect. 2019, 8, 1314–1323. [Google Scholar] [CrossRef] [Green Version]
- Yapa, W.B.; Digana, P.M.C.B.; Ratnasooriya, W.D.; Rübsamen, R.; Costa, H.H.; Randeniya, P.V. Breeding Associated Migration of Miniopterus Schreibersii between Two Natural Caves in Sri Lanka. In Proceedings of the 18th Annual Sessions of the Institute of Biology, Colombo, Sri Lanka, 25–27 September 1998; p. 21. [Google Scholar]
- Yapa, W.B.; Ratnasooriya, W.D. Ecology and Biology of Sri Lankan Bats. Univ. Colombo Rev. 2006, 1, 63–85. [Google Scholar]
- Kusuminda, T.; Mannakkara, A.; Karunarathna, M.; Amarasinghe, C.; Patterson, B.D.; Yapa, W.B. Review of the Distribution of Sri Lankan Bats: Family Miniopteridae. Wildlanka 2020, 8, 152–162. [Google Scholar]
- Tong, S.; Chern, S.W.W.; Li, Y.; Pallansch, M.A.; Anderson, L.J. Sensitive and Broadly Reactive Reverse Transcription-PCR Assays to Detect Novel Paramyxoviruses. J. Clin. Microbiol. 2008, 46, 2652–2658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de Novo Short Read Assembly Using de Bruijn Graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic Trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Genus | March 2018 | June 2018 | January 2019 | Total Urine Samples |
|---|---|---|---|---|
| Miniopterus | 0/0 | 10/102 | 0/11 | 10/113 |
| Rousettus | 0/2 | 0/2 | 0/6 | 0/10 |
| Hipposideros | 0/2 | 0/0 | 0/6 | 0/8 |
| Rhinolophus | 0/6 | 0/0 | 0/6 | 0/12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muzeniek, T.; Perera, T.; Siriwardana, S.; Bayram, F.; Bas, D.; Öruc, M.; Becker-Ziaja, B.; Perera, I.; Weerasena, J.; Handunnetti, S.; et al. Paramyxovirus Diversity within One Population of Miniopterus fuliginosus Bats in Sri Lanka. Pathogens 2022, 11, 434. https://doi.org/10.3390/pathogens11040434
Muzeniek T, Perera T, Siriwardana S, Bayram F, Bas D, Öruc M, Becker-Ziaja B, Perera I, Weerasena J, Handunnetti S, et al. Paramyxovirus Diversity within One Population of Miniopterus fuliginosus Bats in Sri Lanka. Pathogens. 2022; 11(4):434. https://doi.org/10.3390/pathogens11040434
Chicago/Turabian StyleMuzeniek, Therese, Thejanee Perera, Sahan Siriwardana, Fatimanur Bayram, Dilara Bas, Mizgin Öruc, Beate Becker-Ziaja, Inoka Perera, Jagathpriya Weerasena, Shiroma Handunnetti, and et al. 2022. "Paramyxovirus Diversity within One Population of Miniopterus fuliginosus Bats in Sri Lanka" Pathogens 11, no. 4: 434. https://doi.org/10.3390/pathogens11040434
APA StyleMuzeniek, T., Perera, T., Siriwardana, S., Bayram, F., Bas, D., Öruc, M., Becker-Ziaja, B., Perera, I., Weerasena, J., Handunnetti, S., Schwarz, F., Premawansa, G., Premawansa, S., Yapa, W., Nitsche, A., & Kohl, C. (2022). Paramyxovirus Diversity within One Population of Miniopterus fuliginosus Bats in Sri Lanka. Pathogens, 11(4), 434. https://doi.org/10.3390/pathogens11040434