
An Insight into the Genome of Pathogenic and Non-Pathogenic Acanthamoeba


Abstract
1. Introduction
2. Materials and Methods
2.1. Extraction and Sequencing of DNA
2.2. Illumina Genome Assembly
2.3. Hybrid Genome Assembly
2.4. Genome Annotation
3. Results
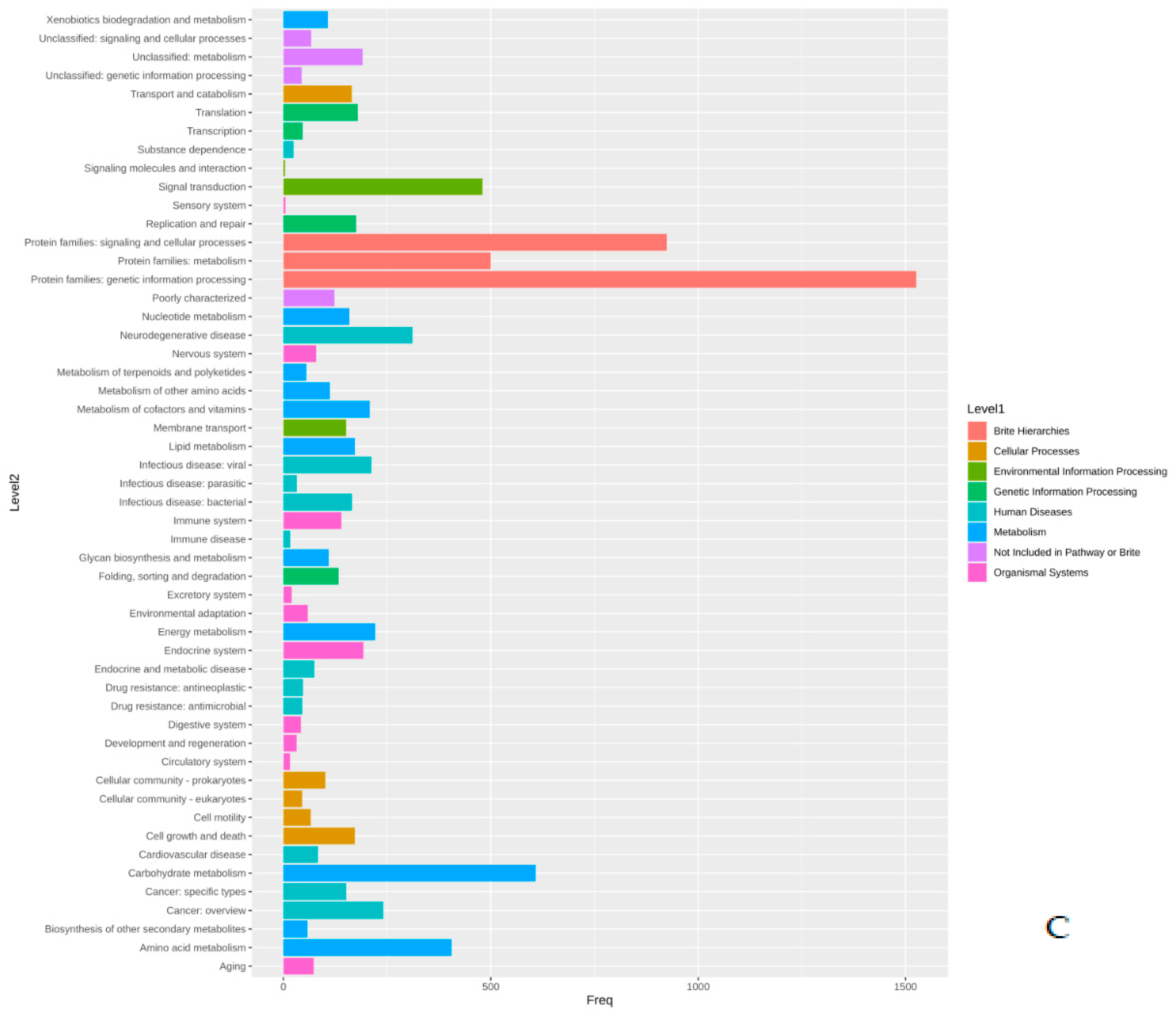
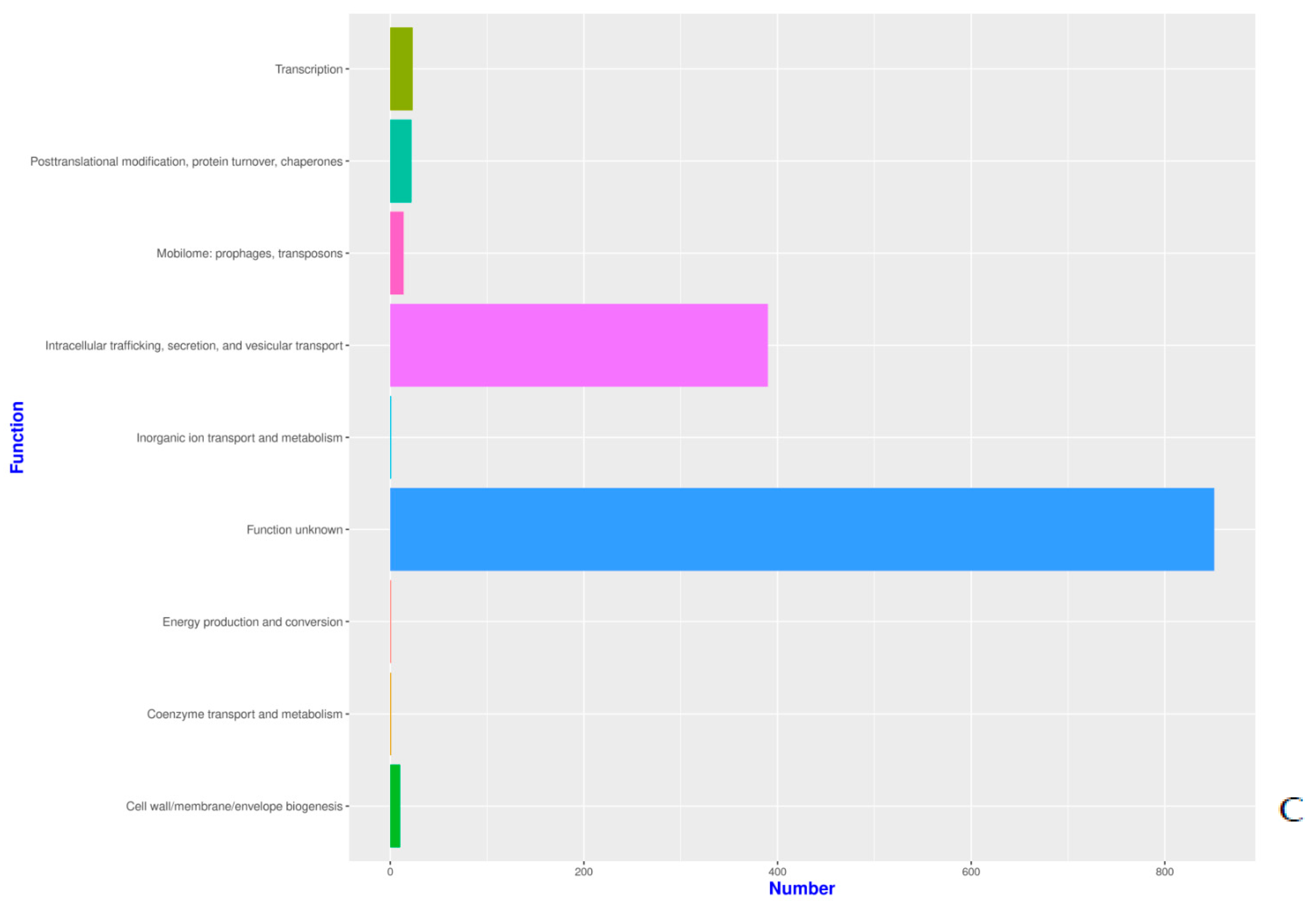
Functional Annotation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kot, K.; Lanocha-Arendarczyk, N.A.; Kosik-Bogacka, D.I. Amoebas from the genus Acanthamoeba and their pathogenic properties. Ann. Parasitol. 2018, 64, 299–308. [Google Scholar] [PubMed]
- Taher, E.E.; Méabed, E.M.H.; Abdallah, I.; Wahed, W.Y.A. Acanthamoeba keratitis in noncompliant soft contact lenses users: Genotyping and risk factors, a study from Cairo, Egypt. J. Infect. Public Health 2018, 11, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Corsaro, D.; Walochnik, J.; Köhsler, M.; Rott, M. Acanthamoeba misidentification and multiple labels: Redefining genotypes T16, T19, and T20 and proposal for Acanthamoeba micheli sp. nov. (genotype T19). Parasitol. Res. 2015, 114, 2481–2490. [Google Scholar] [CrossRef] [PubMed]
- Ertabaklar, H.; Turk, M.; Dayanir, V.; Ertug, S.; Walochnik, J. Acanthamoeba keratitis due to Acanthamoeba genotype T4 in a non-contact-lens wearer in Turkey. Parasitol. Res. 2007, 100, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Ledee, D.R.; Iovieno, A.; Miller, D.; Mandal, N.; Diaz, M.; Fell, J.; Fini, M.E.; Alfonso, E.C. Molecular Identification of T4 and T5 Genotypes in Isolates from Acanthamoeba Keratitis Patients. J. Clin. Microbiol. 2009, 47, 1458–1462. [Google Scholar] [CrossRef]
- Siddiqui, R.; Khan, N.A. Biology and pathogenesis of Acanthamoeba. Parasites Vectors 2012, 5, 6. [Google Scholar] [CrossRef]
- Megha, K.; Sehgal, R.; Khurana, S. Genotyping of Acanthamoeba spp. isolated from patients with granulomatous amoebic encephalitis. Indian J. Med. Res. 2018, 148, 456. [Google Scholar]
- Megha, K.; Sharma, M.; Gupta, A.; Sehgal, R.; Khurana, S. Microbiological diagnosis of Acanthamoebic keratitis: Experience from tertiary care center of North India. Diagn. Microbiol. Infect. Dis. 2021, 100, 115339. [Google Scholar] [CrossRef]
- Megha, K.; Sharma, M.; Sharma, C.; Gupta, A.; Sehgal, R.; Khurana, S. Evaluation of in vitro activity of five antimicrobial agents on Acanthamoeba isolates and their toxicity on human corneal epithelium. Eye 2021, 36, 1911–1917. [Google Scholar] [CrossRef]
- Clarke, M.; Lohan, A.J.; Liu, B.; Lagkouvardos, I.; Roy, S.; Zafar, N.; Bertelli, C.; Schilde, C.; Kianianmomeni, A.; Bürglin, T.R.; et al. Genome of Acanthamoeba castellanii highlights extensive lateral gene transfer and early evolution of tyrosine kinase signaling. Genome Biol. 2013, 14, R11. [Google Scholar] [CrossRef]
- Karlyshev, A.V. Remarkable Features of Mitochondrial DNA of Acanthamoeba polyphaga Linc Ap-1, Revealed by Whole-Genome Sequencing. Microbiol. Resour. Announc. 2019, 8, e00430-19. [Google Scholar] [CrossRef] [PubMed]
- Hasni, I.; Andréani, J.; Colson, P.; La Scola, B. Description of Virulent Factors and Horizontal Gene Transfers of Keratitis-Associated Amoeba Acanthamoeba Triangularis by Genome Analysis. Pathogens 2020, 9, 217. [Google Scholar] [CrossRef] [PubMed]
- Chelkha, N.; Jardot, P.; Moussaoui, I.; Levasseur, A.; La Scola, B.; Colson, P. Core gene-based molecular detection and identification of Acanthamoeba species. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gast, R.J.; Ledee, D.R.; Fuerst, P.A.; Byers, T.J. Subgenus Systematics of Acanthamoeba: Four Nuclear 18S rDNA Sequence Types. J. Eukaryot. Microbiol. 1996, 43, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Stothard, D.R.; Schroeder-Diedrich, J.M.; Awwad, M.H.; Gast, R.J.; Ledee, D.; Rodriguez-Zaragoza, S.; Dean, C.L.; Fuerst, P.; Byers, T.J. The Evolutionary History of the Genus Acanthamoeba and the Identification of Eight New 18S rRNA Gene Sequence Types. J. Eukaryot. Microbiol. 1998, 45, 45–54. [Google Scholar] [CrossRef]
- Gast, R.J. Development of an Acanthamoeba-specific reverse dot-blot and the discovery of a new ribotype. J. Eukaryot. Microbiol. 2001, 48, 609–615. [Google Scholar]
- Fuerst, P.; Booton, G. Species, Sequence Types and Alleles: Dissecting Genetic Variation in Acanthamoeba. Pathogens 2020, 9, 534. [Google Scholar] [CrossRef]
- Xuan, Y.-H.; Chung, B.-S.; Hong, Y.-C.; Kong, H.-H.; Hahn, T.-W.; Chung, D.-I. Keratitis by Acanthamoeba triangularis: Report of Cases and Characterization of Isolates. Korean J. Parasitol. 2008, 46, 157–164. [Google Scholar] [CrossRef]
- Sharma, C.; Thakur, A.; Bhatia, A.; Gupta, A.; Khurana, S. Acanthamoeba keratitis in a mouse model using a novel approach. Indian J. Med. Microbiol. 2021, 39, 523–527. [Google Scholar] [CrossRef]
- Mirjalali, H.; Niyyati, M.; Abedkhojasteh, H.; Babaei, Z.; Sharifdini, M.; Rezaeian, M. Pathogenic Assays of Acanthamoeba Belonging to the T4 Genotype. Iran. J. Parasitol. 2013, 8, 530–535. [Google Scholar]
- Omaña-Molina, M.; Hernandez-Martinez, D.; Sanchez-Rocha, R.; Cardenas-Lemus, U.; Salinas-Lara, C.; Mendez-Cruz, A.R.; Colin-Barenque, L.; Aley-Medina, P.; Espinosa-Villanueva, J.; Moreno-Fierros, L.; et al. In vivo CNS infection model of Acanthamoeba genotype T4: The early stages of infection lack presence of host inflammatory response and are a slow and contact-dependent process. Parasitol. Res. 2017, 116, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef]
- Zimin, A.V.; Puiu, D.; Luo, M.C.; Zhu, T.; Koren, S.; Marçais, G.; Yorke, J.A.; Dvořák, J.; Salzberg, S.L. Hybrid assembly of the large and highly repetitive genome of Aegilops tauschii, a progenitor of bread wheat, with the MaSuRCA mega-reads algorithm. Genome Res. 2017, 27, 787–792. [Google Scholar] [CrossRef]
- Borodovsky, M.; Mills, R.; Besemer, J.; Lomsadze, A. Prokaryotic Gene Prediction Using GeneMark and GeneMark.hmm. Curr. Protoc. Bioinform. 2003, 1, 4.5.1–4.5.16. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35 (Suppl. 2), W182–W185. [Google Scholar] [CrossRef]
- Yang, M.; Derbyshire, M.K.; Yamashita, R.A.; Marchler-Bauer, A. NCBI’s Conserved Domain Database and Tools for Protein Domain Analysis. Curr. Protoc. Bioinform. 2019, 69, e90. [Google Scholar] [CrossRef] [PubMed]
- Oliveros, J.C. VENNY. An Interactive Tool for Comparing Lists with Venn Diagrams. 2007. Available online: http://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 15 September 2022).
- Aurrecoechea, C.; Barreto, A.; Brestelli, J.; Brunk, B.P.; Caler, E.V.; Fischer, S.; Gajria, B.; Gao, X.; Gingle, A.; Grant, G.R.; et al. AmoebaDB and MicrosporidiaDB: Functional genomic resources for Amoebozoa and Microsporidia species. Nucleic Acids Res. 2010, 39, D612–D619. [Google Scholar] [CrossRef] [PubMed]
- Matthey-Doret, C.; Colp, M.J.; Escoll, P.; Thierry, A.; Moreau, P.; Curtis, B.; Sahr, T.; Sarrasin, M.; Gray, M.W.; Lang, B.F.; et al. Chromosome-scale assemblies of Acanthamoeba castellanii genomes provide insights into Legionella pneumophila infection–related chromatin reorganization. Genome Res. 2022, 32, 1698–1710. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Illumina Sequence Data and Quality | ||||||
|---|---|---|---|---|---|---|
| Sample ID | No. of Reads | Data in Gbs | GC % | Read Length | %Q20 | %Q30 |
| SK_2022a | 64,461,292 | 9.67 | 52.5 | 150 | 99.26 | 91.36 |
| SK_2022b | 55,566,470 | 8.34 | 54.5 | 150 | 99.33 | 90.55 |
| SK_2022c | 59,234,380 | 8.89 | 56 | 150 | 99.32 | 93.73 |
| Oxford Nanopore sequence data and quality | ||||||
| Sample ID | No. of sequences | Total length | Average Length | Minimum Length | Maximum Length | |
| SK_2022a | 1,059,703 | 2,724,949,085 | 2,571.40 | 30 | 38,367 | |
| SK_2022b | 1,240,410 | 3,420,889,045 | 2,757.90 | 28 | 38,693 | |
| Assembly | SK_2022a | SK_2022b | SK_2022c |
|---|---|---|---|
| # contigs (>= 0 bp) | 2138 | 1809 | 21,308 |
| # contigs (>= 1000 bp) | 2138 | 1809 | 1634 |
| # contigs (>= 5000 bp) | 1728 | 1666 | 117 |
| # contigs (>= 10000 bp) | 1133 | 1141 | 98 |
| # contigs (>= 25000 bp) | 508 | 527 | 72 |
| # contigs (>= 50000 bp) | 218 | 253 | 46 |
| Largest contig (bp) | 3,411,420 | 2,013,303 | 8,88,447 |
| Total length (bp) | 51,384,221 | 54,719,801 | 22,953,089 |
| GC (%) | 56.73 | 57.1 | 57.91 |
| N50 | 49,572 | 64,210 | 866 |
| N75 | 19,912 | 23,761 | 634 |
| L50 | 222 | 189 | 3186 |
| Alignment % | 79.01% | 76.59% | 93.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, C.; Khurana, S.; Arora, A.; Bhatia, A.; Gupta, A. An Insight into the Genome of Pathogenic and Non-Pathogenic Acanthamoeba. Pathogens 2022, 11, 1558. https://doi.org/10.3390/pathogens11121558
Sharma C, Khurana S, Arora A, Bhatia A, Gupta A. An Insight into the Genome of Pathogenic and Non-Pathogenic Acanthamoeba. Pathogens. 2022; 11(12):1558. https://doi.org/10.3390/pathogens11121558
Chicago/Turabian StyleSharma, Chayan, Sumeeta Khurana, Amit Arora, Alka Bhatia, and Amit Gupta. 2022. "An Insight into the Genome of Pathogenic and Non-Pathogenic Acanthamoeba" Pathogens 11, no. 12: 1558. https://doi.org/10.3390/pathogens11121558
APA StyleSharma, C., Khurana, S., Arora, A., Bhatia, A., & Gupta, A. (2022). An Insight into the Genome of Pathogenic and Non-Pathogenic Acanthamoeba. Pathogens, 11(12), 1558. https://doi.org/10.3390/pathogens11121558