
Detection and Molecular Characterization of Canine Distemper Virus in Wildlife from Northern Italy
 , , , ,
, , , ,  , , ,
, , ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
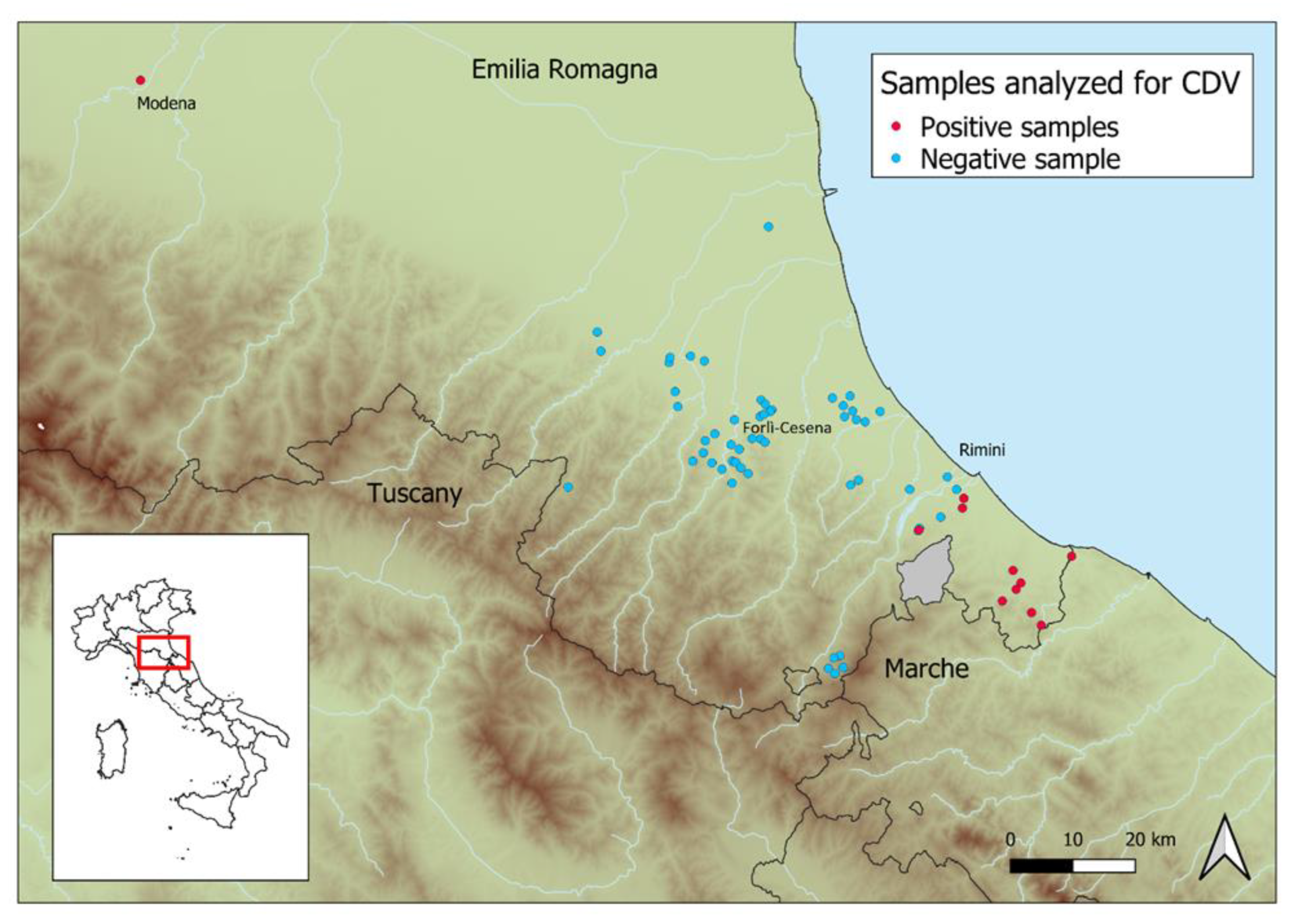
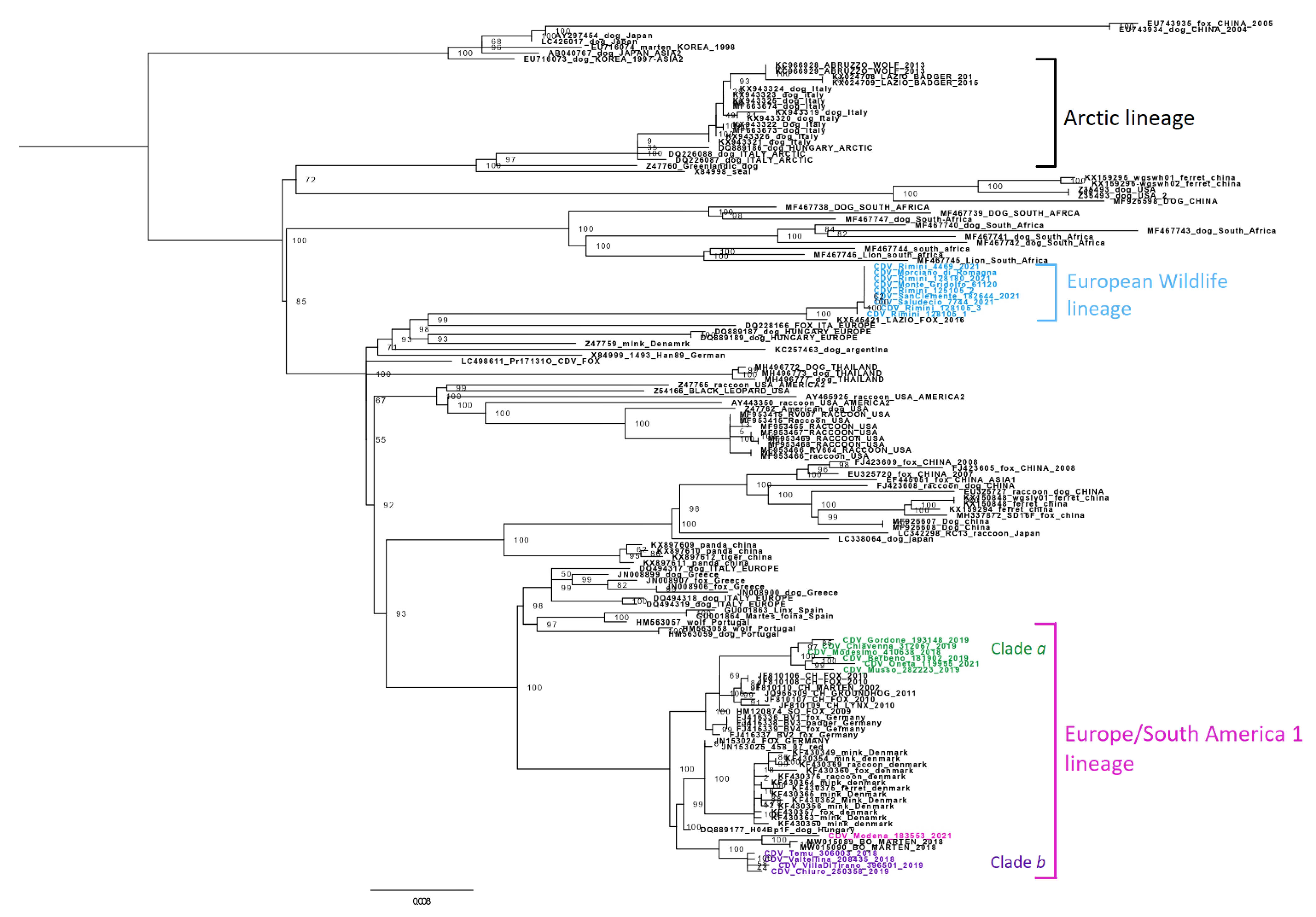
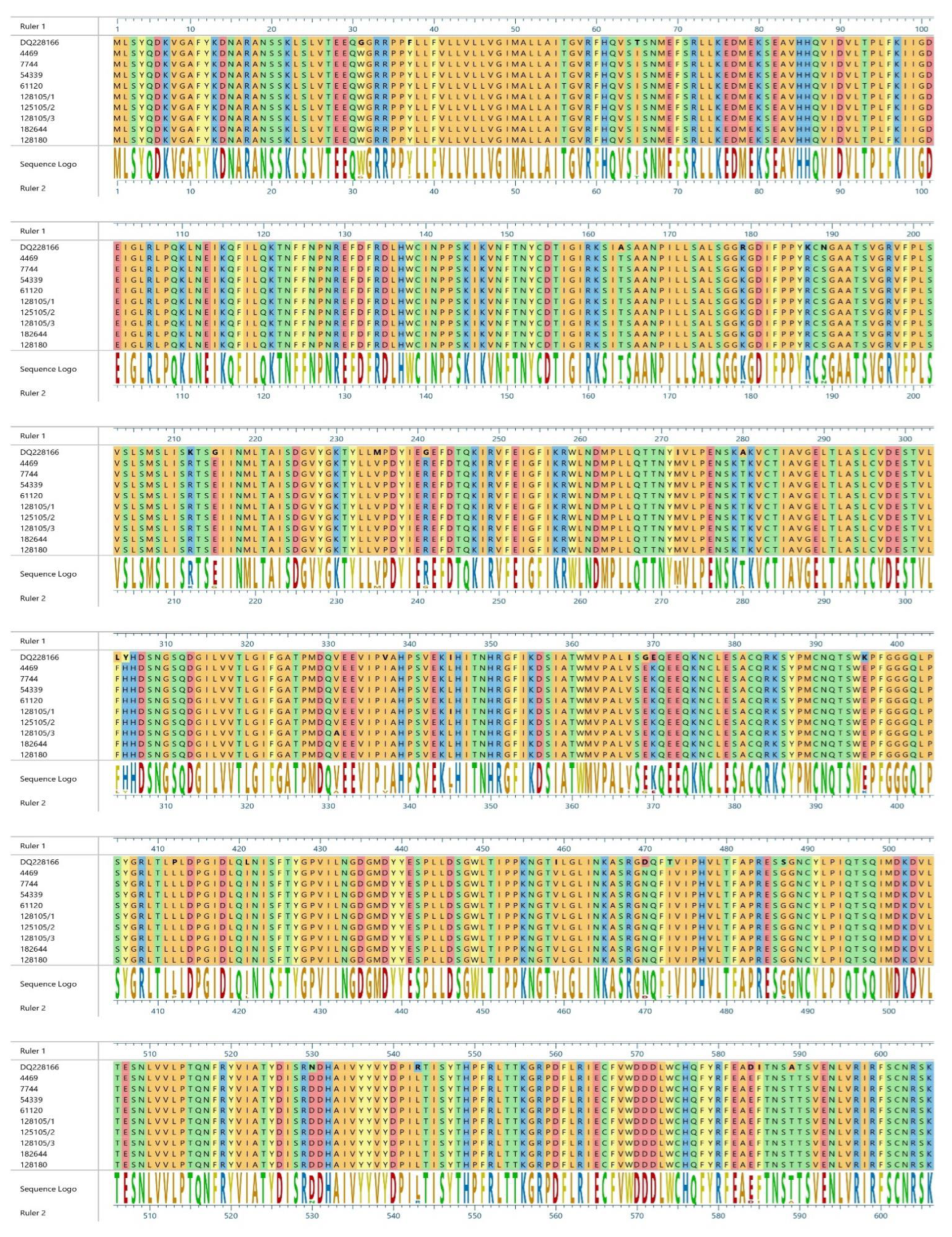
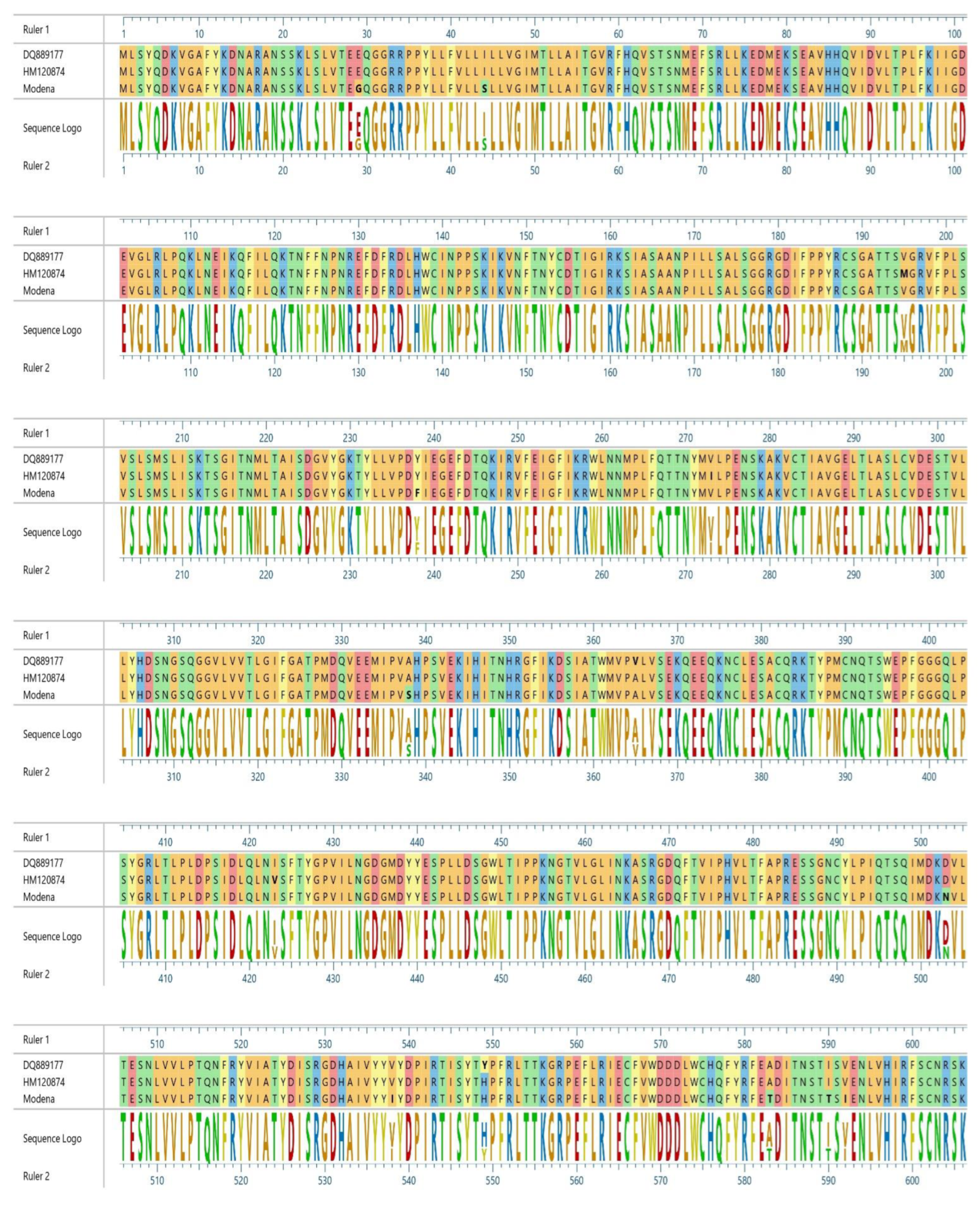
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martinez-Gutierrez, M.; Ruiz-Saenz, J. Diversity of susceptible hosts in canine distemper virus infection: A systematic review and data synthesis. BMC Vet. Res. 2016, 12, 78. [Google Scholar] [CrossRef] [PubMed]
- van Regenmortel, H.V.M.; Fauquet, C.M.; Bishop, D.H.L. Virus taxonomy. In Seventh report of the International Committee on Taxonomy of Viruses; Academic Press: New York, NY, USA, 2000; pp. 556–557. [Google Scholar]
- Tatsuo, H.; Yanagi, Y. The morbillivirus receptor SLAM (CD150). Microbiol. Immunol. 2002, 46, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Noyce, R.S.; Bondre, D.G.; Ha, M.N.; Lin, L.-T.; Sisson, G.; Tsao, M.-S.; Richardson, C.D. Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog. 2011, 7, e1002240. [Google Scholar] [CrossRef]
- Pomeroy, L.W.; Bjornstad, O.N.; Holmes, E.C. The evolutionary and epidemiological dynamics of the paramyxoviridae. J. Mol. Evol. 2008, 66, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Panzera, Y.; Sarute, N.; Iraola, G.; Hernández, M.; Pérez, R. Molecular phylogeography of canine distemper virus: Geographic origin and global spreading. Mol. Phylogenet. Evol. 2015, 92, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wang, X.; Zhai, J.; Zhang, P.; Irwin, D.M.; Shen, X.; Chen, W.; Shen, Y. A new canine distemper virus lineage identified from red pandas in China. Transbound. Emerg. Dis. 2021, 69, e944–e952. [Google Scholar] [CrossRef]
- Monne, I.; Fusaro, A.; Valastro, V.; Citterio, C.; Dalla Pozza, M.; Obber, F.; Trevisiol, K.; Cova, M.; De Benedictis, P.; Bregoli, M.; et al. A distinct CDV genotype causing a major epidemic in Alpine wildlife. Vet. Microbiol. 2011, 150, 63–69. [Google Scholar] [CrossRef]
- Lorusso, A.; Savini, G. Old diseases for new nightmares: Distemper strikes back in Italy. Vet. Ital. 2014, 50, 151–154. [Google Scholar]
- Bianco, A.; Zecchin, B.; Fusaro, A.; Schivo, A.; Ormelli, S.; Bregoli, M.; Citterio, C.V.; Obber, F.; Dellamaria, D.; Trevisiol, K.; et al. Two waves of canine distemper virus showing different spatio-temporal dynamics in Alpine wildlife (2006–2018). Infect. Genet. Evol. 2020, 84, 104359. [Google Scholar] [CrossRef]
- Di Sabatino, D.; Lorusso, A.; Di Francesco, C.E.; Gentile, L.; Di Pirro, V.; Bellacicco, A.L.; Giovannini, A.; Di Francesco, G.; Marruchella, G.; Marsilio, F.; et al. Arctic lineage-canine distemper virus as a cause of death in Apennine wolves (Canis lupus) in Italy. PLoS ONE 2014, 9, e82356. [Google Scholar] [CrossRef]
- Di Sabatino, D.; Di Francesco, G.; Zaccaria, G.; Malatesta, D.; Brugnola, L.; Marcacci, M.; Portanti, O.; De Massis, F.; Savini, G.; Teodori, L.; et al. Lethal distemper in badgers (Meles meles) following epidemic in dogs and wolves. Infect. Genet Evol. 2016, 46, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Balboni, A.; Savini, F.; Scagliarini, A.; Berti, E.; Naldi, M.; Urbani, L.; Fontana, M.C.; Carra, E.; Gibelli, R.M.L.; Gobbo, F.; et al. Natural distemper infection in stone martens (Martes foina): From infection to neutralizing antibodies. Res. Vet. Sci. 2021, 138, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Trogu, T.; Canziani, S.; Salvato, S.; Bianchi, A.; Bertoletti, I.; Gibelli, L.R.; Alborali, G.L.; Barbieri, I.; Gaffuri, A.; Sala, G.; et al. Canine Distemper Outbreaks in Wild Carnivores in Northern Italy. Viruses 2021, 13, 99. [Google Scholar] [CrossRef] [PubMed]
- Rikula, U.; Nuotio, L.; Sihvonen, L. Vaccine coverage, herd immunity and occurrence of canine distemper from 1990–1996 in Finland. Vaccine 2007, 25, 7994–7998. [Google Scholar] [CrossRef]
- Martella, V.; Elia, G.; Buonavoglia, C. Canine distemper virus. Vet. Clin. Small Anim. 2008, 38, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Demeter, Z.; Lakatos, B.; Palade, E.A.; Kozma, T.; Forgách, P.; Rusvai, M. Genetic diversity of Hungarian canine distemper virus strains. Vet. Microbiol. 2007, 122, 258–269. [Google Scholar] [CrossRef]
- Sekulin, K.; Hafner-Marx, A.; Kolodziejek, J.; Janik, D.; Schmidt, P.; Nowotny, N. Emergence of canine distemper in Bavarian wildlife associated with a specific amino acid exchange in the haemagglutinin protein. Vet. J. 2011, 187, 399–401. [Google Scholar] [CrossRef]
- Lednicky, J.A.; Dubach, J.; Kinsel, M.J.; Meehan, T.P.; Bocchetta, M.; Hungerford, L.L.; Sarich, N.A.; Witecki, K.E.; Braid, M.D.; Pedrak, C.; et al. Genetically distant American canine distemper virus lineages have recently caused epizootics with somewhat different characteristics in racoons living around a large suburban zoo in the USA. Virol. J. 2004, 1, 1–14. [Google Scholar] [CrossRef]
- Zhao, J.J.; Yan, X.J.; Chai, X.L.; Martella, V.; Luo, G.L.; Zhang, H.L.; Gao, H.; Liu, Y.; Bai, X.; Zhang, L.; et al. Phylogenetic analysis of the haemagglutinin gene of canine distemper virus strains detected from breeding foxes, raccoon dogs and minks in China. Vet. Microbiol. 2010, 140, 34–42. [Google Scholar] [CrossRef]
- Zhao, J.J.; Zhang, H.L.; Bai, X.; Martella, V.; Hu, B.; Sun, Y.G.; Zhu, C.; Zhang, L.; Liu, H.; Xu, S.; et al. Emergence of canine distemper virus strains with two amino acid substitutions in the haemagglutinin protein, detected from vaccinated carnivores in North-Eastern China in 2012–2013. Vet. J. 2014, 200, 191–194. [Google Scholar] [CrossRef]
- Tao, R.; Chen, J.; Zhao, T.; Gong, C.; Pan, H.; Akhtar, R.W.; Li, X.; Hussain Shah, S.A.; Li, Q.; Zhao, J. Comparison of Growth Characteristics and Genomics of Two Canine Distemper Virus Strains Isolated From Minks in China. Front. Vet. Sci. 2020, 7, 824. [Google Scholar] [CrossRef] [PubMed]
- Origgi, F.C.; Plattet, P.; Sattler, U.; Robert, N.; Casaubon, J.; Mavrot, F.; Pewsner, M.; Wu, N.; Giovannini, S.; Oevermann, A.; et al. Emergence of canine distemper virus strains with modified molecular signature and enhanced neuronal tropism leading to high mortality in wild carnivores. Vet. Pathol. 2012, 49, 913–929. [Google Scholar] [CrossRef] [PubMed]
- WOAH Manual of Diagnostic Tests and Vaccines for Terrestrial Animals 2022, cap. 3.1.18, par B.1.3.1.i “Rabies (Infection with Rebies Virus and Other Lyssaviruses)—Diagnostic Techniques—Identification of the Agent—Laboratory Tests—Immunochemical Identification of Rabies Virus Antigen—Fluorescent Antibody Test”. Available online: https://www.woah.org/fileadmin/Home/eng/Health_standards/tahm/3.01.18_RABIES.pdf (accessed on 1 December 2022).
- WOAH Terrestrial Manual 2018, cap. 3.1.2 par. B.1.2 “Aujeszky’s Disease (Infection with Aujeszky’s Disease Virus)—Diagnostic Techniques- Identification of the Agent—Identification of Virus by the Polymerase Chain Reaction”. Available online: https://www.woah.org/fileadmin/Home/eng/Health_standards/tahm/3.01.02_AUJESZKYS.pdf (accessed on 1 December 2022).
- Rossi, P.; Pozio, E. Guidelines for the detection of Trichinella larvae at the slaughterhouse in a quality assurance system. Ann. Ist. Super Sanità 2008, 44, 195–199. [Google Scholar] [PubMed]
- Galletti, E.; Bonilauri, P.; Bardasi, L.; Fontana, M.C.; Ramini, M.; Renzi, M.; Dosa, G.; Merialdi, G. Development of a minor groove binding probe based real-time PCR for the diagnosis and quantification of Leishmania infantum in dog specimens. Res. Vet. Sci. 2011, 91, 243–245. [Google Scholar] [CrossRef] [PubMed]
- Di Francesco, C.E.; Di Francesco, D.; Di Martino, B.; Speranza, R.; Santori, D.; Boari, A.; Marsilio, F. Detection by hemi-nested reverse transcription polymerase chain reaction and genetic characterization of wild type strains of Canine distemper virus in suspected infected dogs. J. Vet. Diagn. Investig. 2012, 24, 107–115. [Google Scholar] [CrossRef][Green Version]
- Frisk, A.L.; König, M.; Moritz, A.; Baumgärtner, W. Detection of canine distemper virus nucleoprotein RNA by reverse transcription-PCR using serum, whole blood, and cerebrospinal fluid from dogs with distemper. J. Clin. Microbiol. 1999, 37, 3634–3643. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2017, 35, 518–522. [Google Scholar] [CrossRef]
- Pacini, M.I.; Bonelli, F.; Briganti, A.; Citi, S.; Perrucci, S.; Papini, R.A.; Sgorbini, M. Wildlife ungulate rescue and emergency services in the Pisa area (Tuscany, Italy): Evaluation of a 9-years period (2010–2018). Front. Vet. Sci. 2020, 7, 626. [Google Scholar] [CrossRef]
- van Langevelde, F.; van Dooremalen, C.; Jaarsma, C.F. Traffic mortality and the role of minor roads. J. Environ. Manag. 2009, 90, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Fusillo, R.; Romanucci, M.; Marcelli, M.; Massimini, M.; Della Salda, L. Health and Mortality Monitoring in Threatened Mammals: A First Post Mortem Study of Otters (Lutra lutra L.) in Italy. Animals 2022, 12, 609. [Google Scholar] [CrossRef] [PubMed]
- Sleeman, J.M.; Brand, C.J.; Wright, S.D. Strategies for Wildlife Disease Surveillance. Chapter 37 in Applied Techniques of Conservation Medicine 2012; USGS Staff -- Published Research. 971. pp. 539–551. Available online: http://digitalcommons.unl.edu/usgsstaffpub/971 (accessed on 10 December 2022).
- Stallknecht, D.E. Impediments to wildlife disease surveillance, research, and diagnostics. In Wildlife and Emerging Zoonotic Diseases: The Biology, Circumstances and Consequences of Cross-Species Transmission; Childs, J.E., Mackenzie, J.S., Richt, J.A., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 445–461. [Google Scholar]
- Sakai, K.; Nagata, N.; Ami, Y.; Seki, F.; Suzaki, Y.; Iwata-Yoshikawa, N.; Suzuki, T.; Fukushi, S.; Mizutani, T.; Yoshikawa, T.; et al. Lethal canine distemper virus outbreak in cynomolgus monkeys in Japan in 2008. J. Virol. 2013, 87, 1105–1114. [Google Scholar] [CrossRef]
- Pope, J.P.; Miller, D.L.; Riley, M.C.; Anis, E.; Wilkes, R.P. Characterization of a novel canine distemper virus causing disease in wildlife. J. Vet. Diagn. Investig. 2016, 28, 506–513. [Google Scholar] [CrossRef]
- Oleaga, Á.; Vázquez, C.B.; Royo, L.J.; Barral, T.D.; Bonnaire, D.; Armenteros, J.Á.; Rabanal, B.; Gortázar, C.; Balseiro, A. Canine distemper virus in wildlife in south-western Europe. Transbound. Emerg. Dis. 2022, 69, e473–e485. [Google Scholar] [CrossRef]
- Nikolin, V.M.; Wibbelt, G.; Michler, F.U.F.; Wolf, P.; East, M.L. Susceptibility of carnivore hosts to strains of canine distemper virus from distinct genetic lineages. Vet. Microbiol. 2012, 156, 45–53. [Google Scholar] [CrossRef]
- Di Blasio, A.; Irico, L.; Caruso, C.; Miceli, I.; Robetto, S.; Peletto, S.; Varello, K.; Giorda, F.; Mignone, W.; Rubinetti, F.; et al. Canine distemper virus as an emerging multihost pathogen in wild carnivores in northwest Italy. J. Wildl. Dis. 2019, 55, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Ricci, I.; Cersini, A.; Manna, G.; Marcario, G.A.; Conti, R.; Brocherel, G.; Grifoni, G.; Eleni, C.; Scicluna, M.T. A canine distemper virus retrospective study conducted from 2011 to 2019 in central Italy (Latium and Tuscany Regions). Viruses 2021, 13, 272. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PCR steps | Fragments | Primer Sequence |
|---|---|---|
| 1st PCR | 72f | 5′-TACTCTGGTCACACGTCTTA-3′ |
| 798r | 5′-TAGCTCCACTGCATCTGTAT-3′ | |
| 2nd PCR | 472f | 5′-CCGTACATCACCAAGTCATA-3′ |
| 1172r | 5′-TAGAACACCACCTTGTGAAC-3′ | |
| 3rd PCR | 1113f | 5′-GTAGATGAGAGCACCGTATT-3′ |
| 1771r | 5′-TGTGTAGGCAACACCACTAA-3′ | |
| 4th PCR | 1629f | 5′-GGAGACCAGTTCACTGTAAT-3′ |
| 2221r | 5′-GATGGACCTCAGGATATAGA-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trogu, T.; Castelli, A.; Canziani, S.; Tolini, C.; Carrera, M.; Sozzi, E.; Lelli, D.; Tosi, G.; Fiorentini, L.; Di Donato, A.; et al. Detection and Molecular Characterization of Canine Distemper Virus in Wildlife from Northern Italy. Pathogens 2022, 11, 1557. https://doi.org/10.3390/pathogens11121557
Trogu T, Castelli A, Canziani S, Tolini C, Carrera M, Sozzi E, Lelli D, Tosi G, Fiorentini L, Di Donato A, et al. Detection and Molecular Characterization of Canine Distemper Virus in Wildlife from Northern Italy. Pathogens. 2022; 11(12):1557. https://doi.org/10.3390/pathogens11121557
Chicago/Turabian StyleTrogu, Tiziana, Anna Castelli, Sabrina Canziani, Clara Tolini, Maya Carrera, Enrica Sozzi, Davide Lelli, Giovanni Tosi, Laura Fiorentini, Alessandra Di Donato, and et al. 2022. "Detection and Molecular Characterization of Canine Distemper Virus in Wildlife from Northern Italy" Pathogens 11, no. 12: 1557. https://doi.org/10.3390/pathogens11121557
APA StyleTrogu, T., Castelli, A., Canziani, S., Tolini, C., Carrera, M., Sozzi, E., Lelli, D., Tosi, G., Fiorentini, L., Di Donato, A., Rugna, G., Lanci, D., Lavazza, A., & Moreno, A. (2022). Detection and Molecular Characterization of Canine Distemper Virus in Wildlife from Northern Italy. Pathogens, 11(12), 1557. https://doi.org/10.3390/pathogens11121557