
Genomic Variability of SARS-CoV-2 Omicron Variant Circulating in the Russian Federation during Early December 2021 and Late January 2022

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
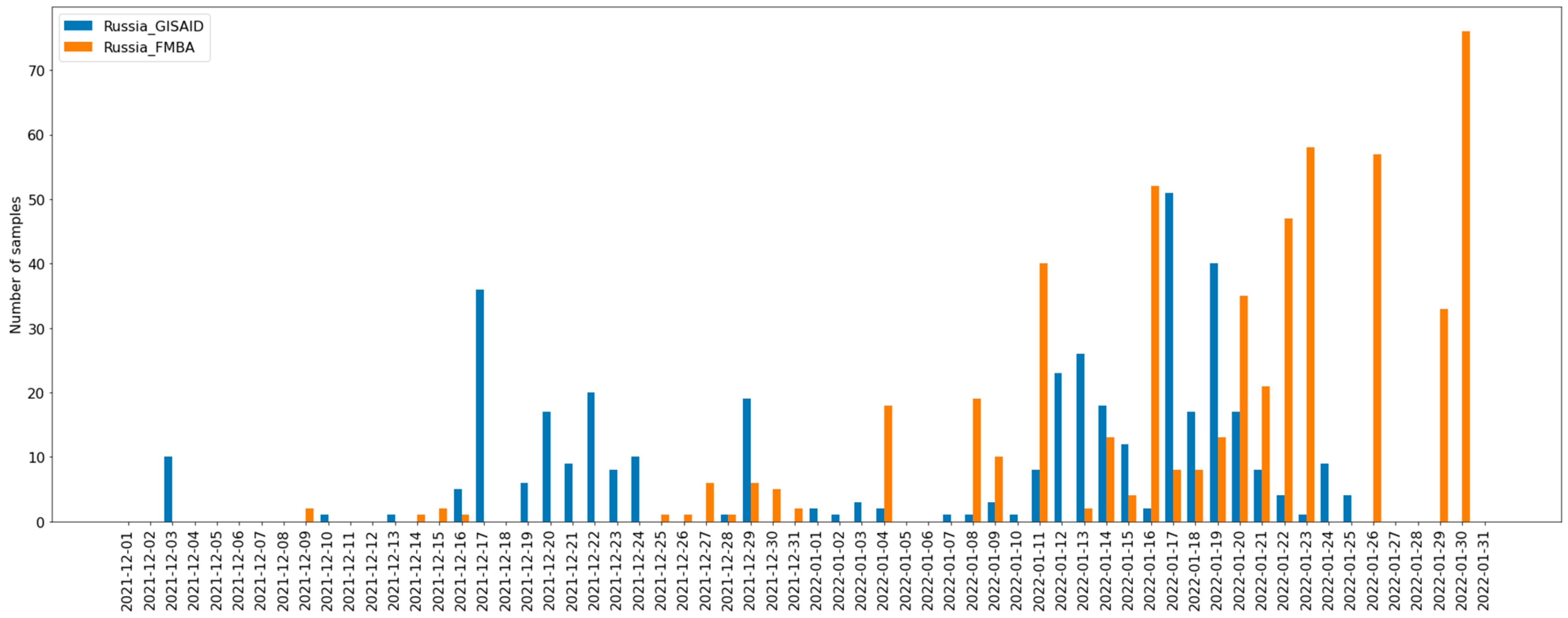
2.1. Sample Collection
2.2. Whole Genome Sequencing
2.3. Consensus Calling
2.4. Data Preparation and Phylogenetic Analysis
3. Results
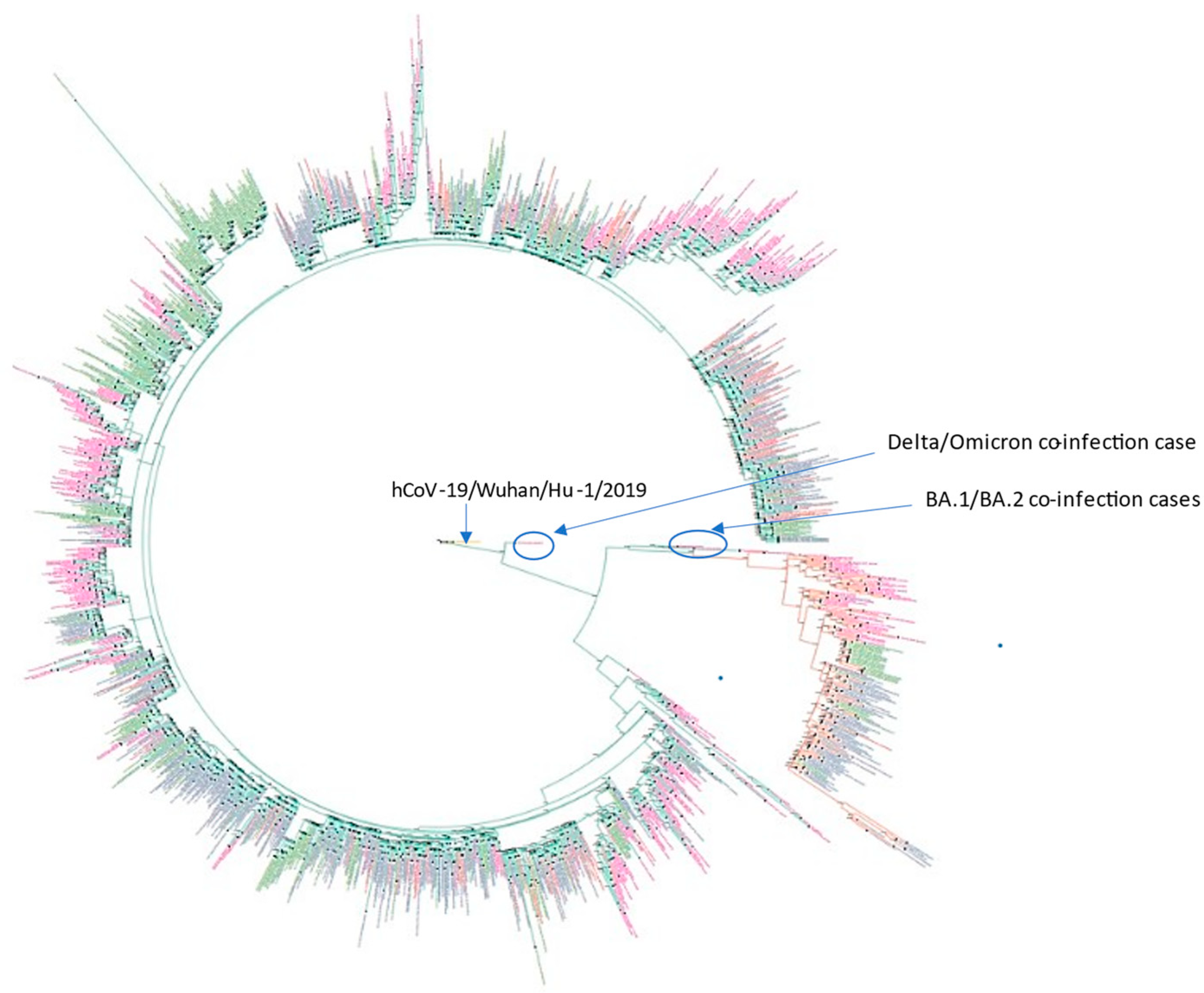
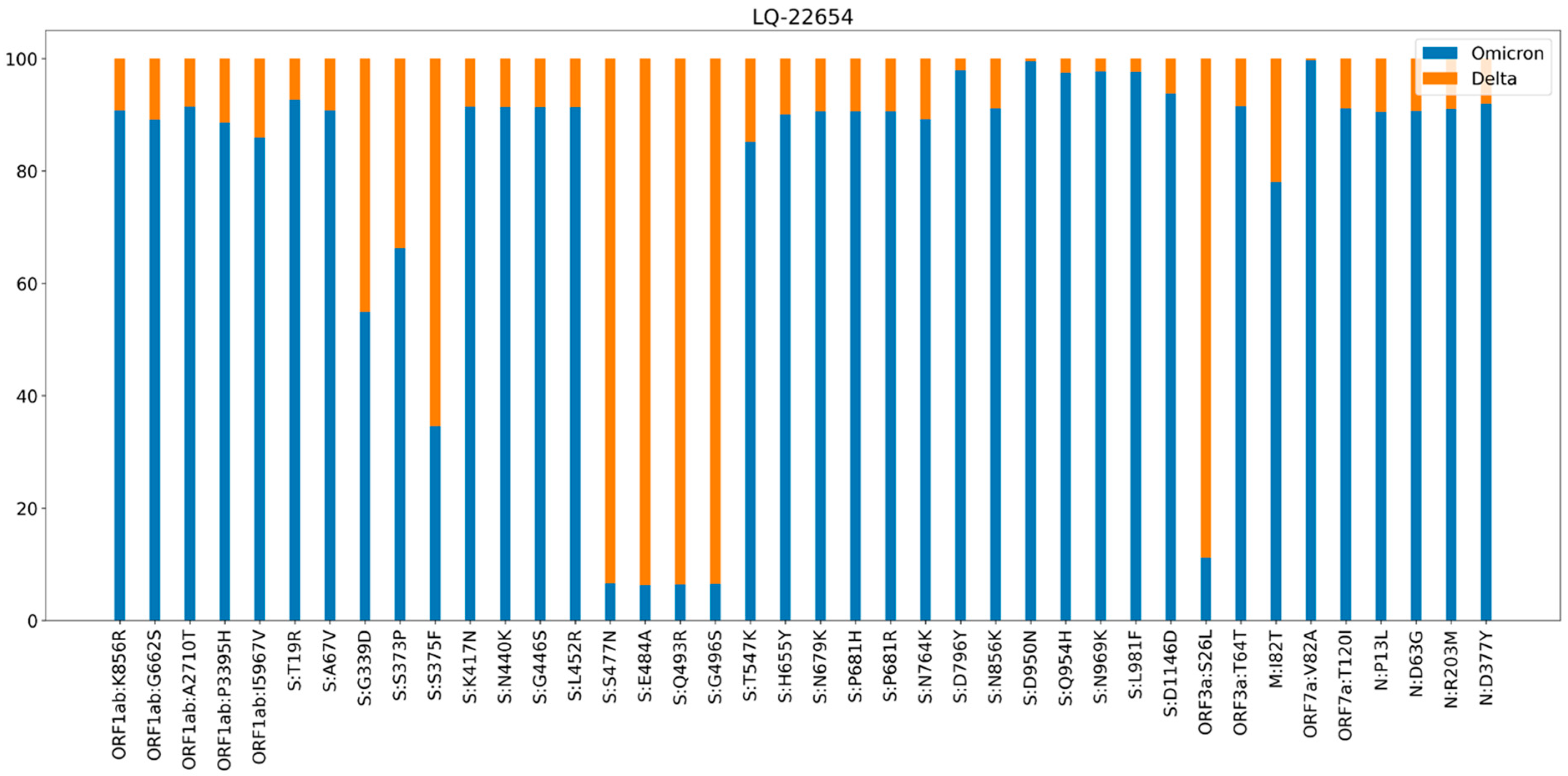
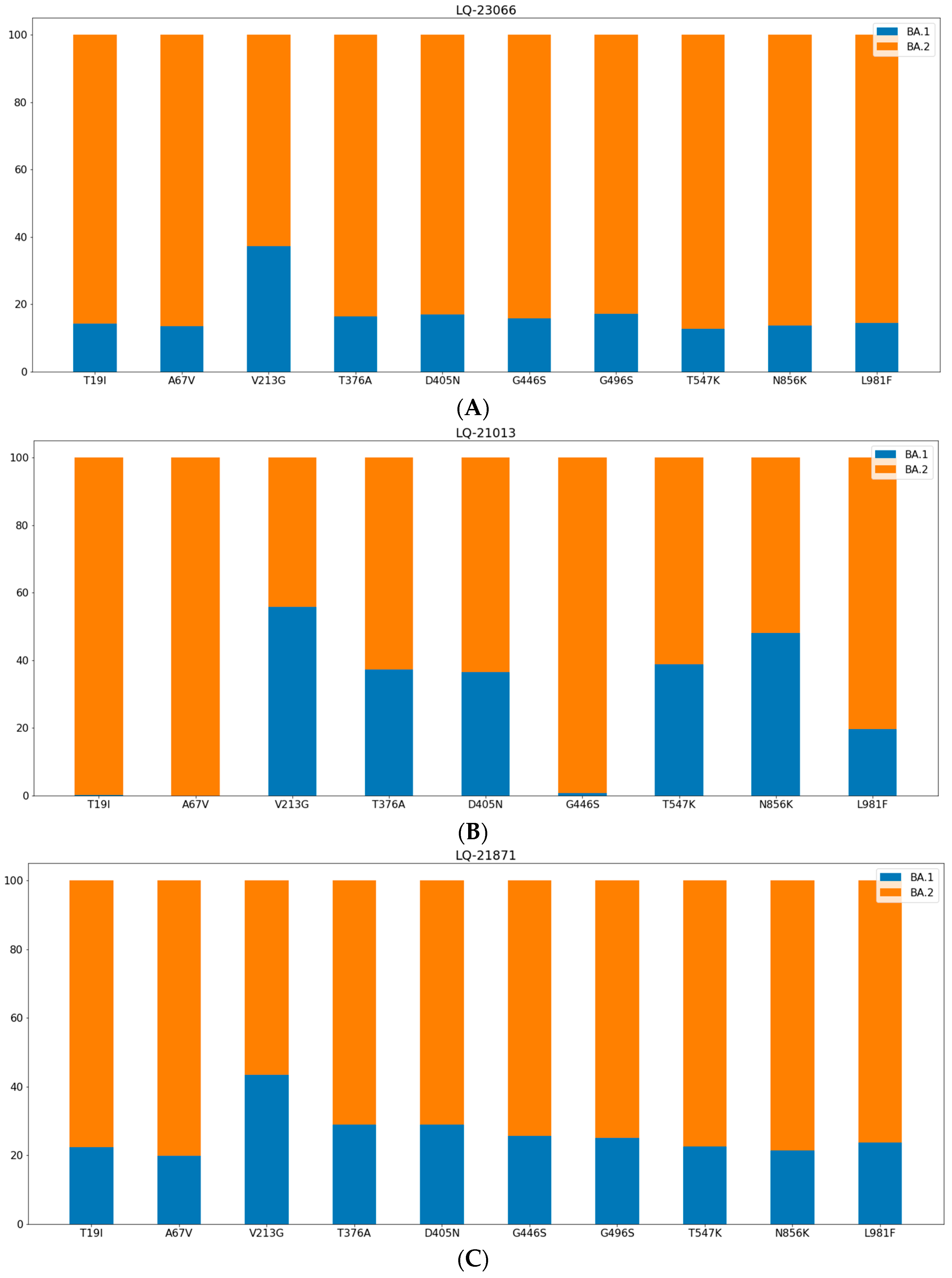
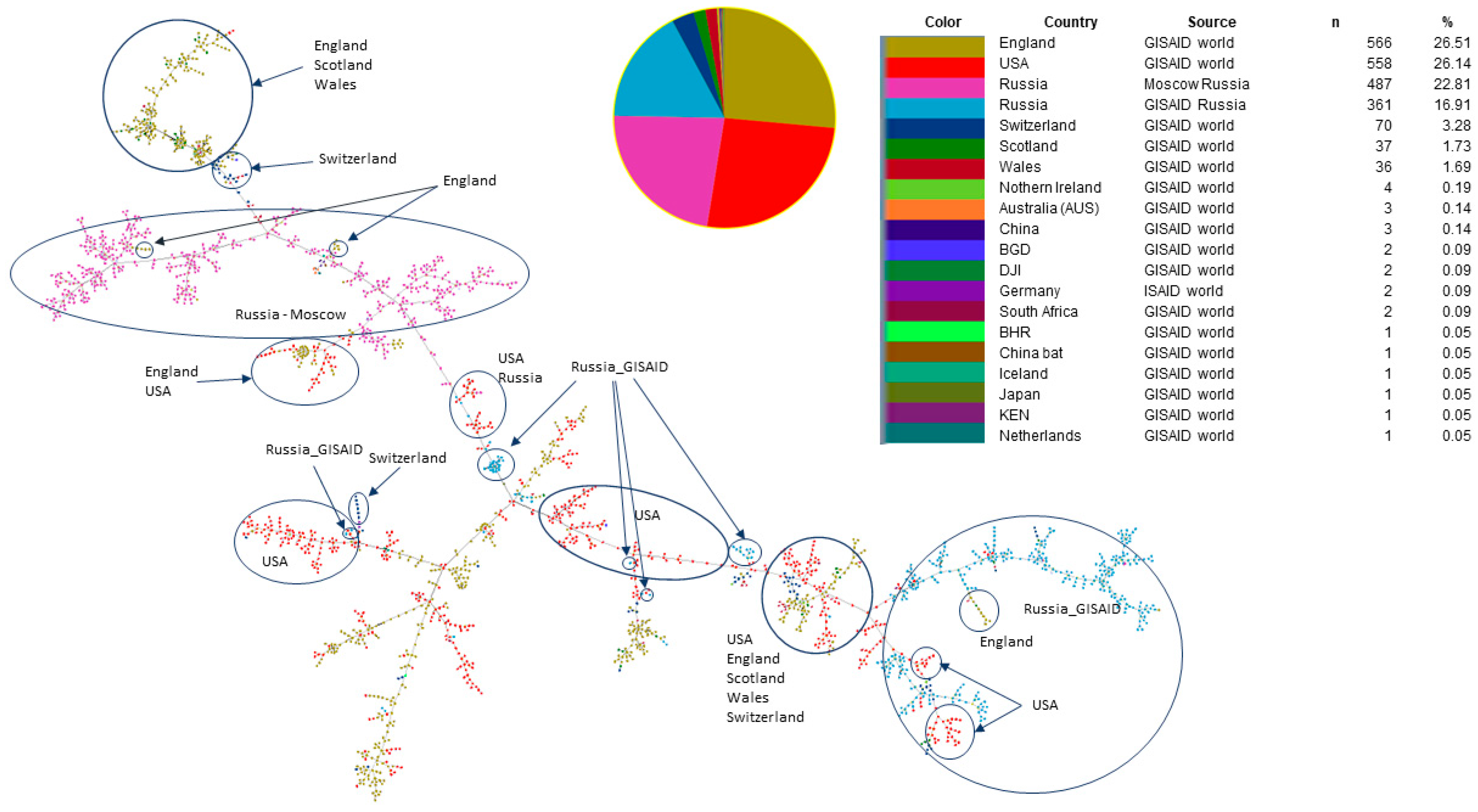
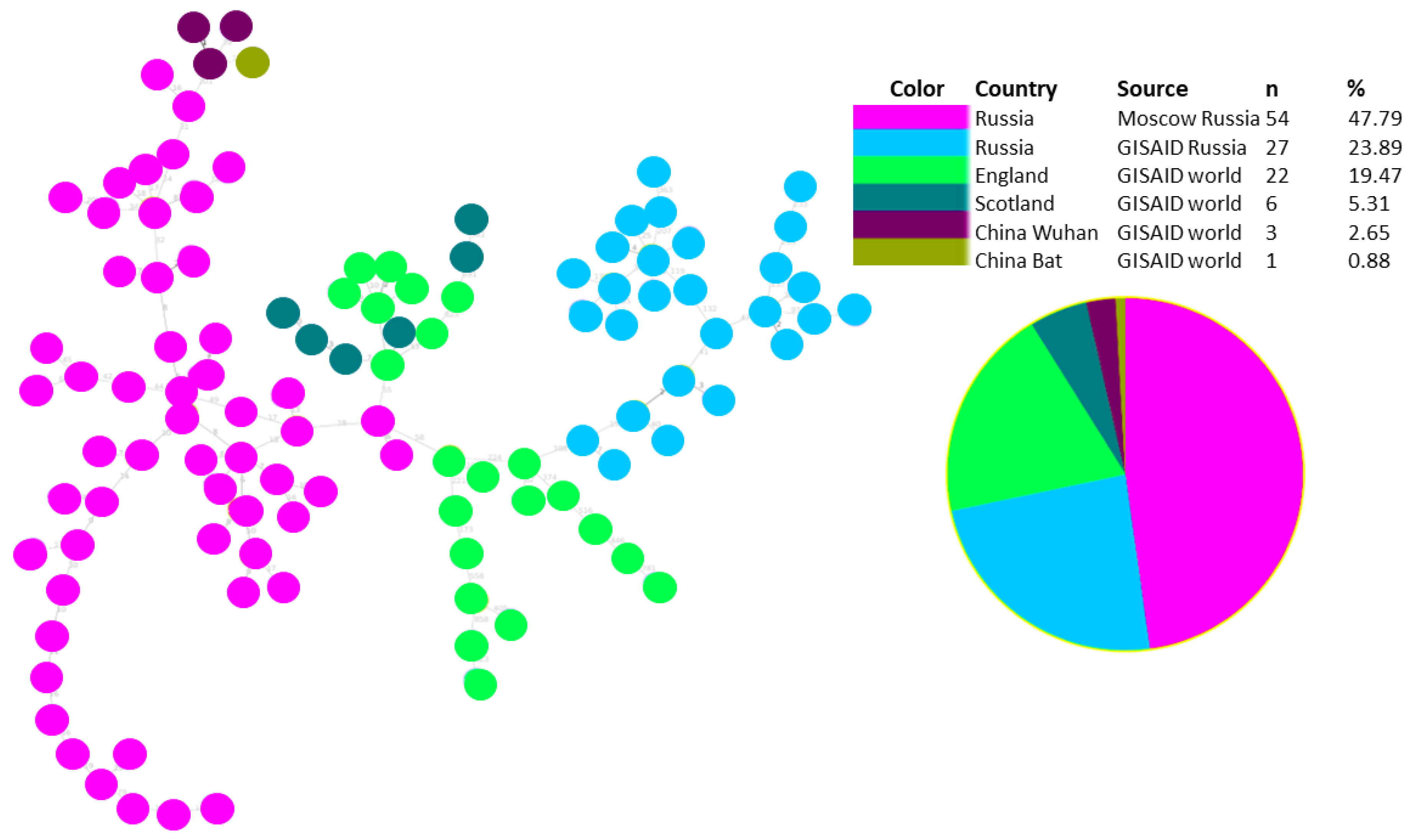
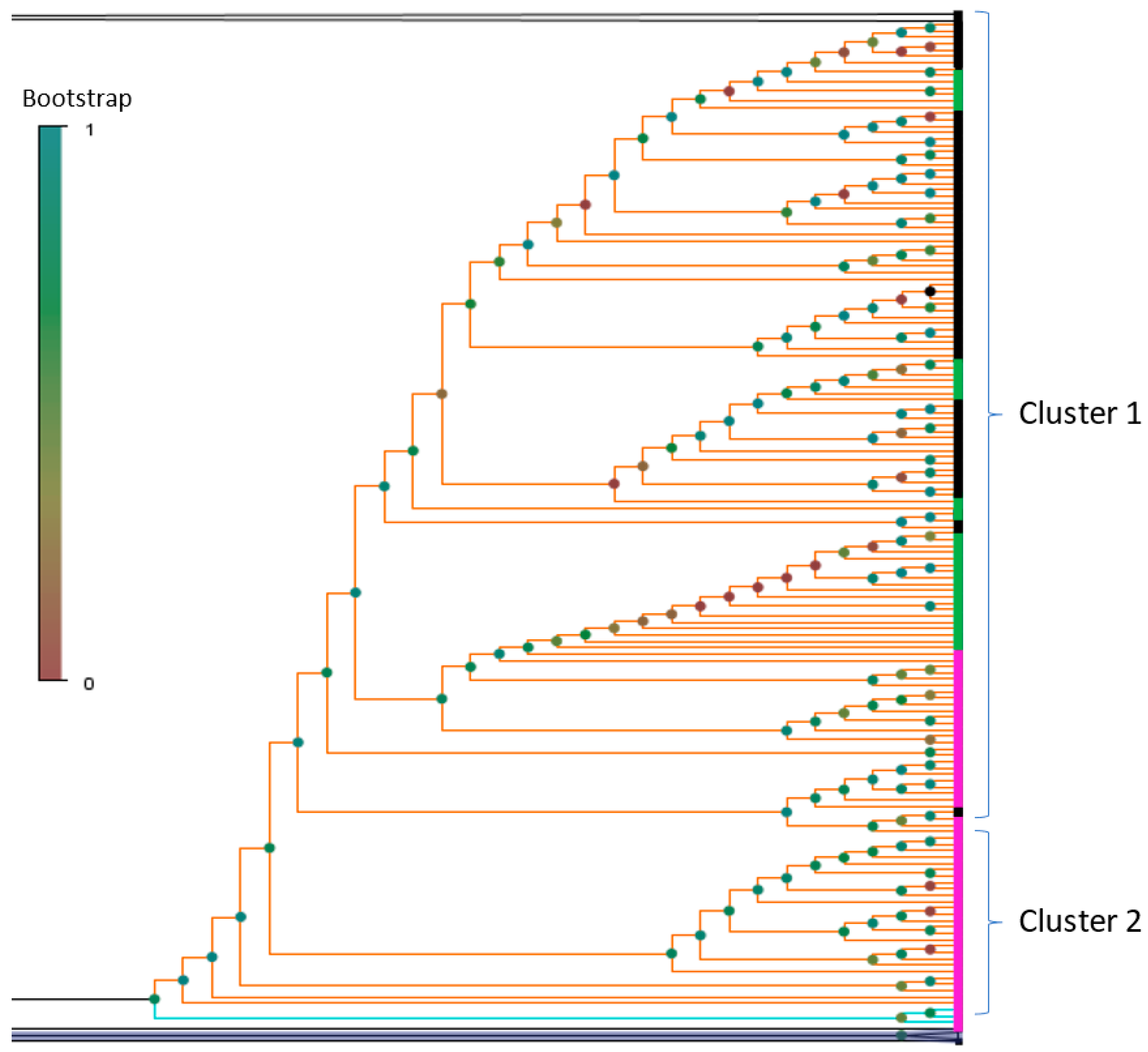
3.1. Phylogenetic Analysis and Coinfection Identification
- samples LQ-23066 and LQ-21871 were from different PCR plates,
- sample LQ-22654 did not have any Delta strains in the closest wells or in the same row of the plate,
- there were no other samples from the same plates that contained sequencing reads from various genomes (no other contamination/coinfection cases were identified).
3.2. Investigation of the SARS-CoV-2 Omicron Variant Origin in Russia
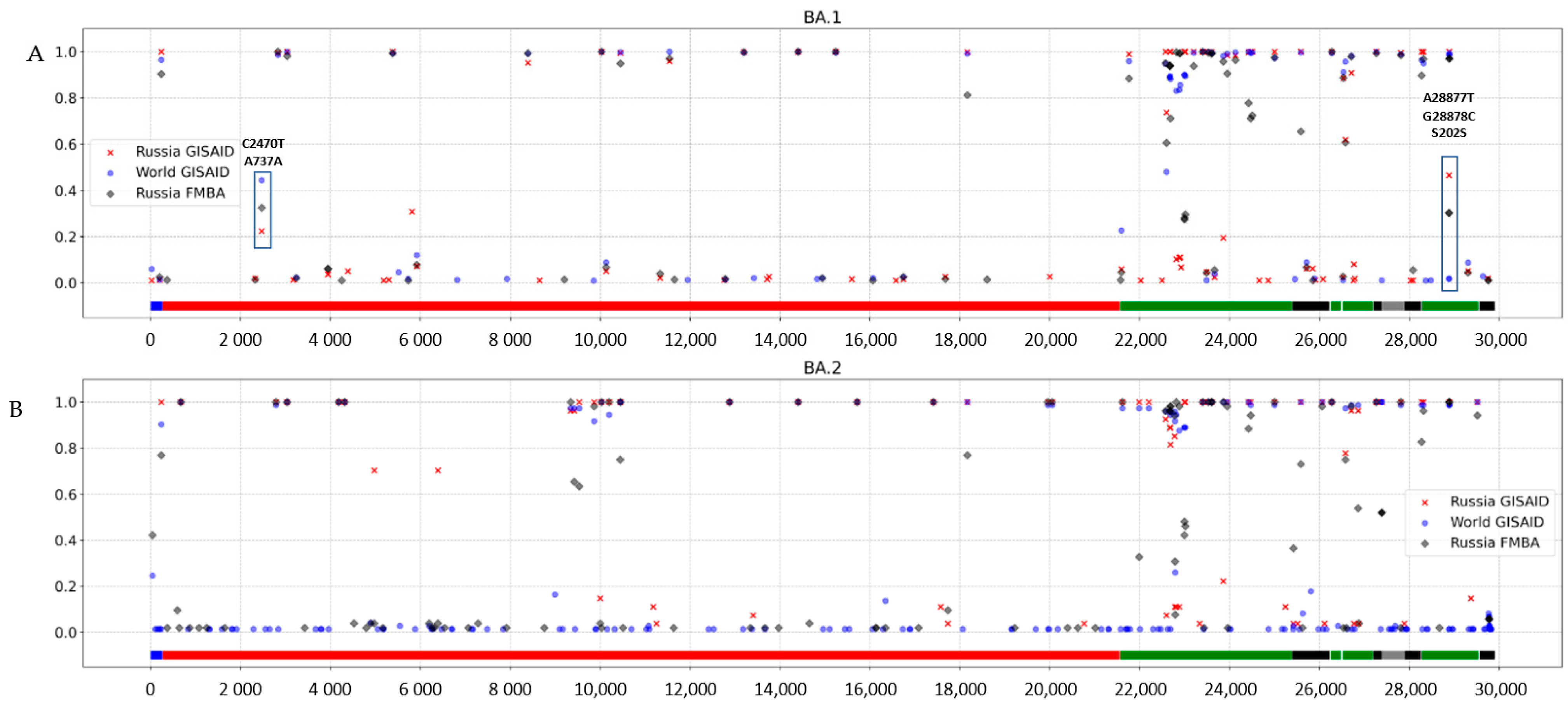
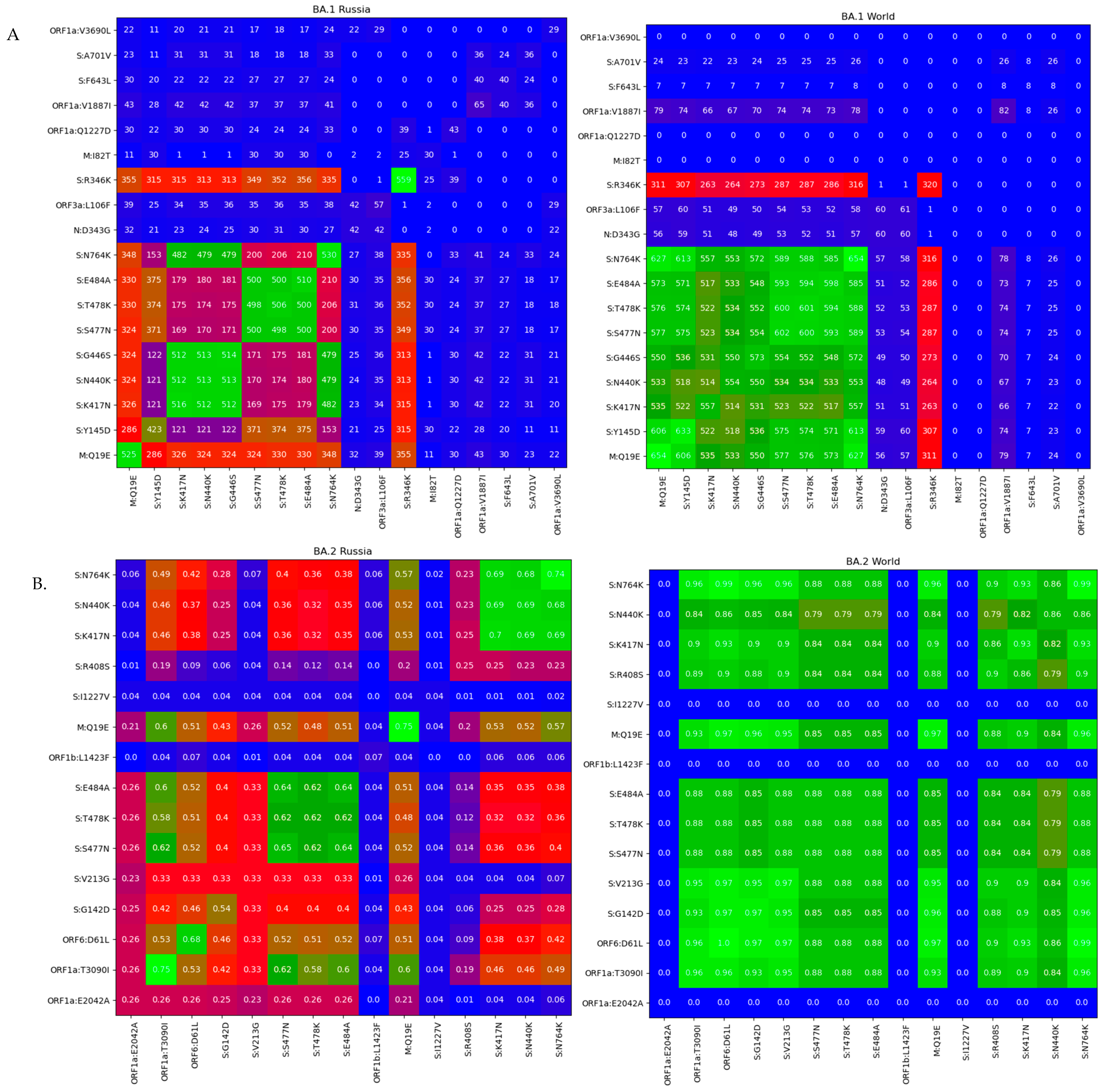
3.3. Genomic Variation across Russian Omicron Strains
4. Discussion
- convergent evolution of different viral lineages and the appearance of mutations that provide advantages from the point of view of evolution;
- coinfection cases, where a set of nontypical major viral variant mutations were introduced into the consensus sequence due to specificsequencing procedures or assembly pipeline features;
- recombination events that can lead to appearance of nontypical combination of mutations in the genome;
- contamination causing unusual combinations of substitutions.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Classification of Omicron (B.1.1.529): SARS-CoV-2 Variant of Concern. Available online: https://www.who.int/news/item/26-11-2021-classification-of-omicron-(b.1.1.529)-sars-cov-2-variant-of-concern (accessed on 1 June 2022).
- B.1.1.529. Available online: https://cov-lineages.org/global_report_B.1.1.529.html (accessed on 1 June 2022).
- Phylogenetic Analysis of SARS-CoV-2 Clusters in Their International Context-Cluster 21K.Omicron. Available online: https://nextstrain.org/groups/neherlab/ncov/21K.Omicron?label=clade:21M%20%28Omicron%29 (accessed on 1 June 2022).
- Kupferschmidt, K. Where did “weird” Omicron come from? Science 2021, 374, 1179. [Google Scholar] [CrossRef] [PubMed]
- Statement on Omicron Sublineage BA.2-WHO. Available online: https://www.who.int/news/item/22-02-2022-statement-on-omicron-sublineage-ba.2 (accessed on 1 August 2022).
- Song, Z.; Xu, Y.; Bao, L.; Zhang, L.; Yu, P.; Qu, Y.; Zhu, H.; Zhao, W.; Han, Y.; Qin, C. From SARS to MERS, Thrusting Coronaviruses into the Spotlight. Viruses 2019, 11, 59. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Kandeel, M.; Mohamed, M.E.M.; Abd El-Lateef, H.M.; Venugopala, K.N.; El-Beltagi, H.S. Omicron variant genome evolution and phylogenetics. J. Med. Virol. 2022, 94, 1627–1632. [Google Scholar] [CrossRef] [PubMed]
- Omicron Variant: What You Need to Know. Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/omicron-variant.html (accessed on 1 August 2022).
- Wang, L.; Cheng, G. Sequence analysis of the emerging SARS-CoV-2 variant Omicron in South Africa. J. Med. Virol. 2022, 94, 1728–1733. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- PRINSEQ. Available online: http://prinseq.sourceforge.net (accessed on 10 May 2022).
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Nextclade. Available online: https://docs.nextstrain.org/projects/nextclade/en/stable/user/nextclade-cli.html (accessed on 10 May 2022).
- FastTree 2.1.11. Available online: http://www.microbesonline.org/fasttree/ (accessed on 10 May 2022).
- Turakhia, Y.; Thornlow, B.; Hinrichs, A.S.; De Maio, N.; Gozashti, L.; Lanfear, R.; Haussler, D.; Corbett-Detig, R. Ultrafast Sample placement on Existing tRees (UShER) enables real-time phylogenetics for the SARS-CoV-2 pandemic. Nat. Genet. 2021, 53, 809–816. [Google Scholar] [CrossRef]
- Ribeiro-Gonçalves, B.; Francisco, A.P.; Vaz, C.; Ramirez, M.; Carriço, J.A. PHYLOViZ Online: Web-based tool for visualization, phylogenetic inference, analysis and sharing of minimum spanning trees. Nucleic Acids Res. 2016, 44, W246–W251. [Google Scholar] [CrossRef]
- Colson, P.; Delerce, J.; Beye, M.; Levasseur, A.; Boschi, C.; Houhamdi, L.; Tissot-Dupont, H.; Yahi, N.; Million, M.; La Scola, B.; et al. First cases of infection with the 21L/BA.2 Omicron variant in Marseille, France. J. Med. Virol. 2022, 94, 3421–3430. [Google Scholar] [CrossRef]
- SARS-CoV-2 Variant Classifications and Definitions. Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-classifications.html (accessed on 1 August 2022).
- Feil, E.J.; Li, B.C.; Aanensen, D.M.; Hanage, W.P.; Spratt, B.G. eBURST: Inferring Patterns of Evolutionary Descent among Clusters of Related Bacterial Genotypes from Multilocus Sequence Typing Data. J. Bacteriol. 2004, 186, 1518–1530. [Google Scholar] [CrossRef]
- Francisco, A.P.; Bugalho, M.; Ramirez, M.; Carriço, J.A. Global optimal eBURST analysis of multilocus typing data using a graphic matroid approach. BMC Bioinform. 2009, 10, 152. [Google Scholar] [CrossRef]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef]
- Combes, P.; Bisseux, M.; Bal, A.; Marin, P.; Archimbaud, C.; Brebion, A.; Chabrolles, H.; Regagnon, C.; Lafolie, J.; Destras, G.; et al. Evidence of co-infection during Delta and Omicron variants of concern co-circulation, weeks 49-2021 to 02-2022, France. medRxiv 2022. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, T.; Fang, Y.; Liu, J.; Ye, Q.; Ding, L. SARS-CoV-2 spike L452R mutation increases Omicron variant fusogenicity and infectivity as well as host glycolysis. Sig. Transduct. Target Ther. 2022, 7, 76. [Google Scholar] [CrossRef]
- Bolze, A.; Basler, T.; White, S.; Rossi, A.D.; Wyman, D.; Roychoudhury, P.; Greninger, A.L.; Hayashibara, K.; Beatty, M.; Shah, S.; et al. Evidence for SARS-CoV-2 Delta and Omicron co-infections and recombination. medRxiv 2022. [Google Scholar] [CrossRef]
- Syed, A.M.; Taha, T.Y.; Tabata, T.; Chen, I.P.; Ciling, A.; Khalid, M.M.; Sreekumar, B.; Chen, P.-Y.; Hayashi, J.M.; Soczek, K.M.; et al. Rapid assessment of SARS-CoV-2–evolved variants using virus-like particles. Science 2021, 374, 1626–1632. [Google Scholar] [CrossRef]
- Flemming, A. Omicron, the great escape artist. Nat. Rev. Immunol. 2022, 22, 75. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, J.; Jian, F.; Xiao, T.; Song, W.; Yisimayi, A.; Huang, W.; Li, Q.; Wang, P.; An, R.; et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature 2022, 602, 657–663. [Google Scholar] [CrossRef]
- Peacock, T.P.; Brown, J.C.; Zhou, J.; Thakur, N.; Newman, J.; Kugathasan, R.; Yan, A.W.; Furnon, W.; De Lorenzo, G.; Cowton, V.M.; et al. The SARS-CoV-2 Variant, Omicron, Shows Rapid Replication in Human Primary Nasal Epithelial Cultures and Efficiently Uses the Endosomal Route of Entry. bioRxiv 2022. [Google Scholar] [CrossRef]
- Pulliam, J.R.C.; van Schalkwyk, C.; Govender, N.; von Gottberg, A.; Cohen, C.; Groome, M.J.; Dushoff, J.; Mlisana, K.; Moultrie, H. Increased risk of SARS-CoV-2 reinfection associated with emergence of Omicron in South Africa. medRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Lewnard, J.A.; Hong, V.X.; Patel, M.M.; Kahn, R.; Lipsitch, M.; Tartof, S.Y. Clinical Outcomes Among Patients Infected with Omicron (B.1.1.529) SARS-CoV-2 Variant in Southern California. medRxiv 2022. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | Number of Samples |
|---|---|
| FMBA | 542 |
| GISAID global | 756 |
| GISAID Russian | 397 |
| UShER | 1702 |
| Wuhan genomes | 4 |
| Bat coronavirus RaTG13 | 1 |
| Group of Strains | Number of BA.1 Strains | Number of BA.2 Strains | Number of Non-Clustered Strains |
|---|---|---|---|
| Russian Moscow | 483 | 55 | 4 |
| Russian GISAID | 370 | 27 | 0 |
| Global sample GISAID | 683 | 73 | 0 |
| Total | 1536 | 155 | 4 |
| Substitution | Number of Genomes | Pangolin Classification |
|---|---|---|
| M:I82T | 18 | BA.1, BA.1.1., BA.1.1.13, BA.1.1.15, BA.2 |
| S:L452R | 12 | BA.1, BA.1.1, BA.1.17 |
| S:L452R, M:I82T | 12 | BA.1, BA.1.1, BA.1.17, |
| N:R203M | 1 | BA.1.1 |
| S:L452R, M:I82T, ORF1a:P2046L | 1 | BA.1.1 |
| Type of Epitope | BA.1 Mutations | BA.2 Mutations | Neutralizing Epitopes in BA.1 and BA.2 | Total |
|---|---|---|---|---|
| Discontinuous Epitopes Number | 228 | 196 | 82 | 311 |
| Linear Epitopes Number | 3 | 3 | 5 | 8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chernyaeva, E.N.; Ayginin, A.A.; Bulusheva, I.A.; Vinogradov, K.S.; Stetsenko, I.F.; Romanova, S.V.; Tsypkina, A.V.; Matsvay, A.D.; Savochkina, Y.A.; Shipulin, G.A. Genomic Variability of SARS-CoV-2 Omicron Variant Circulating in the Russian Federation during Early December 2021 and Late January 2022. Pathogens 2022, 11, 1461. https://doi.org/10.3390/pathogens11121461
Chernyaeva EN, Ayginin AA, Bulusheva IA, Vinogradov KS, Stetsenko IF, Romanova SV, Tsypkina AV, Matsvay AD, Savochkina YA, Shipulin GA. Genomic Variability of SARS-CoV-2 Omicron Variant Circulating in the Russian Federation during Early December 2021 and Late January 2022. Pathogens. 2022; 11(12):1461. https://doi.org/10.3390/pathogens11121461
Chicago/Turabian StyleChernyaeva, Ekaterina N., Andrey A. Ayginin, Irina A. Bulusheva, Kirill S. Vinogradov, Ivan F. Stetsenko, Svetlana V. Romanova, Anastasia V. Tsypkina, Alina D. Matsvay, Yulia A. Savochkina, and German A. Shipulin. 2022. "Genomic Variability of SARS-CoV-2 Omicron Variant Circulating in the Russian Federation during Early December 2021 and Late January 2022" Pathogens 11, no. 12: 1461. https://doi.org/10.3390/pathogens11121461
APA StyleChernyaeva, E. N., Ayginin, A. A., Bulusheva, I. A., Vinogradov, K. S., Stetsenko, I. F., Romanova, S. V., Tsypkina, A. V., Matsvay, A. D., Savochkina, Y. A., & Shipulin, G. A. (2022). Genomic Variability of SARS-CoV-2 Omicron Variant Circulating in the Russian Federation during Early December 2021 and Late January 2022. Pathogens, 11(12), 1461. https://doi.org/10.3390/pathogens11121461