

Molecular Characteristics and Genetic Evolution of Echovirus 33 in Mainland of China

 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Viral Isolation and Molecular Typing
2.2. Whole-Genome Sequencing of E33
2.3. Dataset Establishment
2.4. Recombination Analysis of the Seven Isolated E33 Strains
2.5. Phylogenetic Analysis of E33
3. Results
3.1. Five Genotypes Were Reclassified Based on the VP1 Sequences
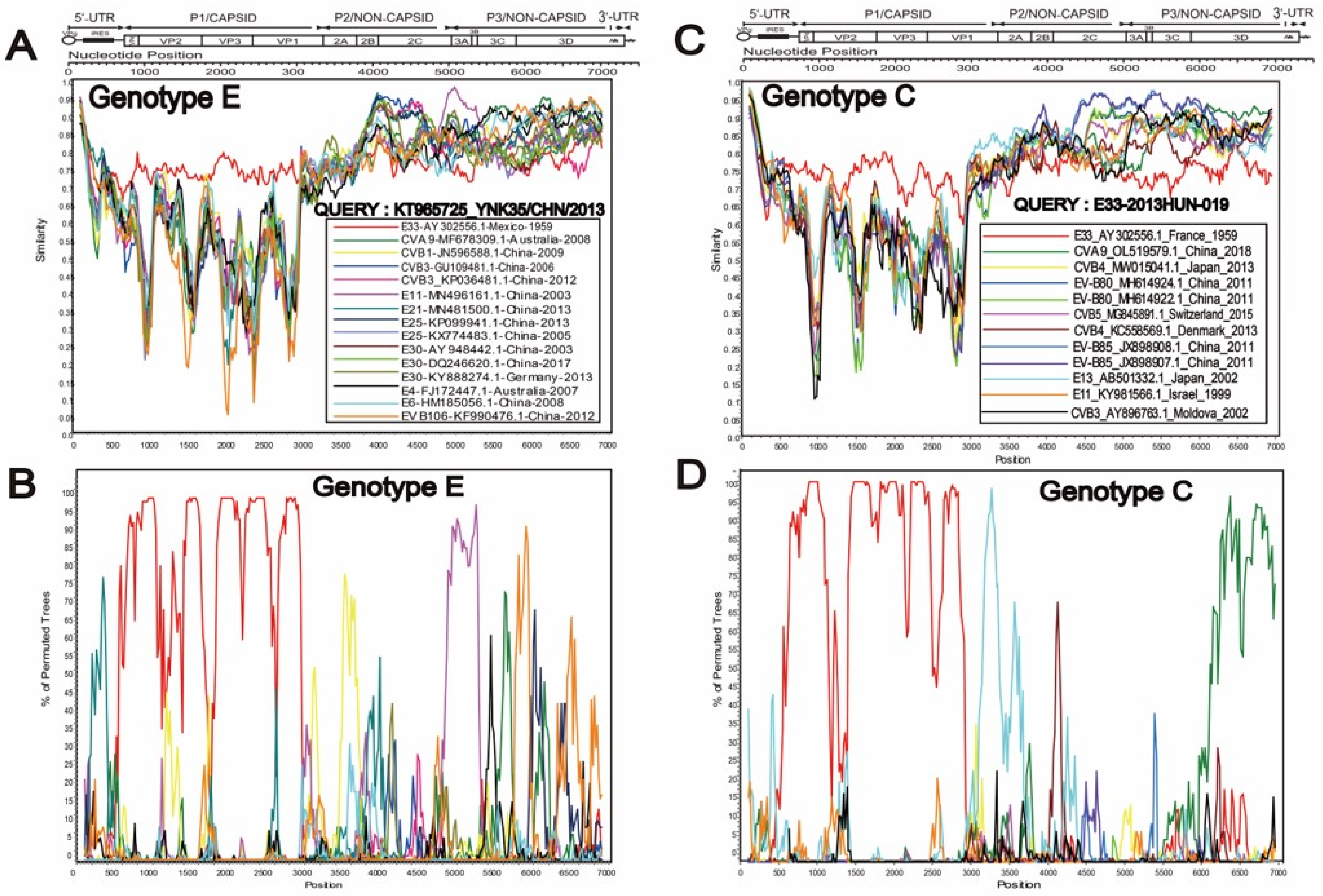
3.2. Two Different Recombination Patterns Were Found between Different Genotypes
3.3. Special Recombination Pattern of Genotype C
3.4. High Nucleotide Substitution Rate in the VP1 Region of E33
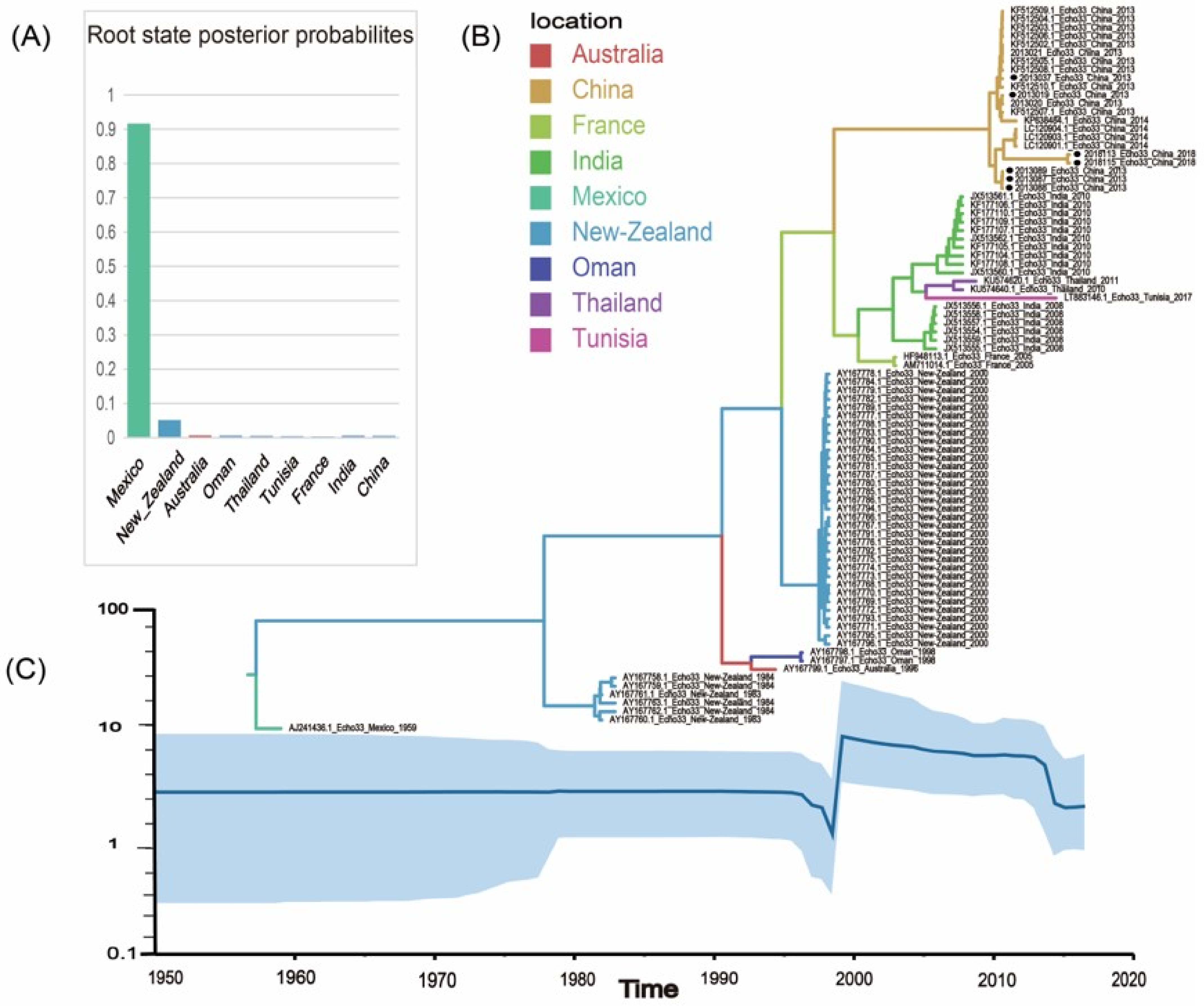
3.5. Global Geographic Transmission Paths of E33
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Davison, A.J.; Dempsey, D.M.; Dutilh, B.E.; García, M.L.; et al. Changes to virus taxonomy and to the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2021). Arch. Virol. 2021, 166, 2633–2648. [Google Scholar] [CrossRef] [PubMed]
- Glenet, M.; Heng, L.; Callon, D.; Lebreil, A.-L.; Gretteau, P.-A.; Nguyen, Y.; Berri, F.; Andreoletti, L. Structures and Functions of Viral 5′ Non-Coding Genomic RNA Domain-I in Group-B Enterovirus Infections. Viruses 2020, 12, 919. [Google Scholar] [CrossRef] [PubMed]
- Pascal, S.M.; Garimella, R.; Warden, M.S.; Ponniah, K. Structural Biology of the Enterovirus Replication-Linked 5′-Cloverleaf RNA and Associated Virus Proteins. Microbiol. Mol. Biol. Rev. 2020, 84. [Google Scholar] [CrossRef] [PubMed]
- Dutkiewicz, M.; Stachowiak, A.; Swiatkowska, A.; Ciesiołka, J. Structure and function of RNA elements present in enteroviral genomes. Acta Biochim. Pol. 2017, 63, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Oberste, M.S.; Maher, K.; Kilpatrick, D.R.; Flemister, M.R.; Brown, B.A.; Pallansch, M.A. Typing of Human Enteroviruses by Partial Sequencing of VP1. J. Clin. Microbiol. 1999, 37, 1288–1293. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhang, Y.; Fan, Q.; Cui, H.; Yan, D.; Zhu, S.; Tang, H.; Sun, Q.; Wang, D.; Xu, W. Phylogenetic Characterizations of Highly Mutated EV-B106 Recombinants Showing Extensive Genetic Exchanges with Other EV-B in Xinjiang, China. Sci. Rep. 2017, 7, 43080. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Zhang, Y.; Hong, M.; Han, Z.; Zhang, M.; Song, Y.; Yan, D.; Zhu, S.; Xu, W. Phylogenetic characteristics and molecular epidemiological analysis of novel enterovirus EV-B83 isolated from Tibet, China. Sci. Rep. 2020, 10, 6630. [Google Scholar] [CrossRef]
- Zhang, Y.; Hong, M.; Sun, Q.; Zhu, S.; Tsewang; Li, X.; Yan, D.; Wang, D.; Xu, W. Molecular typing and characterization of a new serotype of human enterovirus (EV-B111) identified in China. Virus Res. 2014, 183, 75–80. [Google Scholar] [CrossRef]
- Lu, H.; Hong, M.; Zhang, Y.; Xiao, J.; Zhang, M.; Zhang, K.; Song, Y.; Han, Z.; Yang, Q.; Wang, D.; et al. A novel interspecies recombinant enterovirus (Enterovirus A120) isolated from a case of acute flaccid paralysis in China. Emerg. Microbes Infect. 2020, 9, 1733–1743. [Google Scholar] [CrossRef]
- Lukashev, A.N. Role of recombination in evolution of enteroviruses. Rev. Med Virol. 2005, 15, 157–167. [Google Scholar] [CrossRef]
- Muslin, C.; Mac Kain, A.; Bessaud, M.; Blondel, B.; Delpeyroux, F. Recombination in Enteroviruses, a Multi-Step Modular Evolutionary Process. Viruses. 2019, 11, 859. [Google Scholar] [CrossRef] [PubMed]
- Druyts-Voets, E.; Yane, F.; Bosmans, E.; Colaert, J.; Desmyter, J. Method for selecting optimal cells for enterovirus isolation as determined in an outbreak of echovirus type 33 meningitis. Eur. J. Clin. Microbiol. 1985, 4, 331–334. [Google Scholar] [CrossRef] [PubMed]
- Henigst, W. Echovirus type 33 in Southern Germany (Bavaria). Epidemiological studies after isolation of the virus from cerebrospinal fluid. Zentralblatt fur Bakteriol. Parasitenkd. Infekt. und Hyg. 1. Abt. Med. Bakteriol. Virusforsch. und Parasitol. Orig. 1968, 206, 133–139. [Google Scholar]
- Quillien, M.C.; Baruteau, R.; Alix, D.; Lejeune, B.; Jehan, P.; Chastel, C. An epidemic of Echovirus 33 infection in Brittany. Ann. Pediatr. 1983, 30, 409–413. [Google Scholar]
- Sato, K.; Yamashita, T.; Sakae, K.; Suzuki, Y.; Ishikawa, N.; Nishimura, Y. A new-born baby outbreak of echovirus type 33 infection. J. Infect. 1998, 37, 123–126. [Google Scholar] [CrossRef]
- Huang, Q.S.; Carr, J.M.; Nix, W.A.; Oberste, M.S.; Kilpatrick, D.R.; Pallansch, M.A.; Croxson, M.C.; Lindeman, J.A.; Baker, M.G.; Grimwood, K. An Echovirus Type 33 Winter Outbreak in New Zealand. Clin. Infect. Dis. 2003, 37, 650–657. [Google Scholar] [CrossRef][Green Version]
- Grimwood, K.; Huang, Q.S.; Sadleir, L.G.; Nix, W.A.; Kilpatrick, D.R.; Oberste, M.S.; Pallansch, M.A. Acute Flaccid Paralysis from Echovirus Type 33 Infection. J. Clin. Microbiol. 2003, 41, 2230–2232. [Google Scholar] [CrossRef]
- Rao, D.C.; Babu, M.A.; Raghavendra, A.; Dhananjaya, D.; Kumar, S.; Maiya, P. Non-polio enteroviruses and their association with acute diarrhea in children in India. Infect. Genet. Evol. 2013, 17, 153–161. [Google Scholar] [CrossRef]
- Rao, C.D.; Yergolkar, P.; Shankarappa, K.S. Antigenic Diversity of Enteroviruses Associated with Nonpolio Acute Flaccid Paralysis, India, 2007–2009. Emerg. Infect. Dis. 2012, 18, 1833–1840. [Google Scholar] [CrossRef]
- Sadeuh-Mba, S.A.; Bessaud, M.; Massenet, D.; Joffret, M.-L.; Endegue, M.-C.; Njouom, R.; Reynes, J.-M.; Rousset, D.; Delpeyroux, F. High Frequency and Diversity of Species C Enteroviruses in Cameroon and Neighboring Countries. J. Clin. Microbiol. 2013, 51, 759–770. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, H.; Zhao, Y.; Zhang, H.; Sun, H.; Huang, X.; Yang, Z.; Liu, J.; Ma, S. Identification of a new recombinant strain of echovirus 33 from children with hand, foot, and mouth disease complicated by meningitis in Yunnan, China. Virol. J. 2019, 16, 1–7. [Google Scholar] [CrossRef]
- Tan, X.; Gao, L.; Ma, X.; Nie, J.; Zhan, D.; Zhang, B.; Liu, Y.; Liu, F.; Xu, W. An outbreak of echovirus 33 in schools in China in 2013. Arch. Virol. 2014, 159, 2233–2241. [Google Scholar] [CrossRef]
- Yang, C.-F.; Naguib, T.; Yang, S.-J.; Nasr, E.; Jorba, J.; Ahmed, N.; Campagnoli, R.; van der Avoort, H.; Shimizu, H.; Yoneyama, T.; et al. Circulation of Endemic Type 2 Vaccine-Derived Poliovirus in Egypt from 1983 to 1993. J. Virol. 2003, 77, 8366–8377. [Google Scholar] [CrossRef]
- Ishiko, H.; Shimada, Y.; Yonaha, M.; Hashimoto, O.; Hayashi, A.; Sakae, K.; Takeda, N. Molecular Diagnosis of Human Enteroviruses by Phylogeny-Based Classification by Use of the VP4 Sequence. J. Infect. Dis. 2002, 185, 744–754. [Google Scholar] [CrossRef]
- Martin, D.; Posada, D.; Crandall, K.; Williamson, C. A Modified Bootscan Algorithm for Automated Identification of Recombinant Sequences and Recombination Breakpoints. AIDS Res. Hum. Retrovir. 2005, 21, 98–102. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian Phylogeography Finds Its Roots. PLOS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef]
- Xie, W.; Lewis, P.O.; Fan, Y.; Kuo, L.; Chen, M.-H. Improving Marginal Likelihood Estimation for Bayesian Phylogenetic Model Selection. Syst. Biol. 2010, 60, 150–160. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian Coalescent Inference of Past Population Dynamics from Molecular Sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Baele, G.; Lemey, P.; Rambaut, A.; A Suchard, M. Adaptive MCMC in Bayesian phylogenetics: An application to analyzing partitioned data in BEAST. Bioinformatics 2017, 33, 1798–1805. [Google Scholar] [CrossRef] [PubMed]
- Nikolaidis, M.; Mimouli, K.; Kyriakopoulou, Z.; Tsimpidis, M.; Tsakogiannis, D.; Markoulatos, P.; Amoutzias, G.D. Large-scale genomic analysis reveals recurrent patterns of intertypic recombination in human enteroviruses. Virology 2018, 526, 72–80. [Google Scholar] [CrossRef]
- Lukashev, A.N.; Lashkevich, V.A.; Koroleva, G.A.; Ilonen, J.; Hinkkanen, A.E. Recombination in uveitis-causing enterovirus strains. J. Gen. Virol. 2004, 85, 463–470. [Google Scholar] [CrossRef]
- Bouslama, L.; Nasri, D.; Chollet, L.; Belguith, K.; Bourlet, T.; Aouni, M.; Pozzetto, B.; Pillet, S. Natural Recombination Event within the Capsid Genomic Region Leading to a Chimeric Strain of Human Enterovirus B. J. Virol. 2007, 81, 8944–8952. [Google Scholar] [CrossRef][Green Version]
- Vakulenko, Y.; Deviatkin, A.; Lukashev, A. Using Statistical Phylogenetics for Investigation of Enterovirus 71 Genotype A Reintroduction into Circulation. Viruses 2019, 11, 895. [Google Scholar] [CrossRef]
- Zhang, M.; Zhao, Y.; Zhang, H.; Lin, K.; Liu, H.; Zhang, J.; Ding, L.; Huang, X.; Yang, Z.; Ma, S. Molecular characterization of Coxsackievirus A16 strains isolated from children with severe hand, foot, and mouth disease in Yunnan, Southwest China, during 2009-2015. J. Med. Virol. 2018, 91, 155–160. [Google Scholar] [CrossRef]
- Tan, X.; Li, L.; Zhang, B.; Jorba, J.; Su, X.; Dongjing, Y.; Yang, D.; Lv, L.; Li, J.; Xu, W. Molecular epidemiology of coxsackievirus A6 associated with outbreaks of hand, foot, and mouth disease in Tianjin, China, in 2013. Arch. Virol. 2015, 160, 1097–1104. [Google Scholar] [CrossRef]
- Bian, L.; Gao, F.; Mao, Q.; Sun, S.; Wu, X.; Liu, S.; Yang, X.; Liang, Z. Hand, foot, and mouth disease associated with coxsackievirus A10: More serious than it seems. Expert Rev. Anti-infective Ther. 2019, 17, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wang, J.; Chen, J.; Huang, R.; Zhang, Y.; Xiao, J.; Song, Y.; Ji, T.; Yang, Q.; Zhu, S.; et al. Molecular Epidemiology and Evolution of Coxsackievirus A9. Viruses 2022, 14, 822. [Google Scholar] [CrossRef]
- Lukashev, A.N.; Vakulenko, Y.A. Molecular evolution of types in non-polio enteroviruses. J. Gen. Virol. 2017, 98, 2968–2981. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Fan, H.; Zhou, S.; Li, S.; NIGEDELI, A.; Zhang, Y.; Sun, Q.; He, Y.; Guo, Q.; Wang, X.; et al. Molecular Characteristics and Genetic Evolution of Echovirus 33 in Mainland of China. Pathogens 2022, 11, 1379. https://doi.org/10.3390/pathogens11111379
Wang W, Fan H, Zhou S, Li S, NIGEDELI A, Zhang Y, Sun Q, He Y, Guo Q, Wang X, et al. Molecular Characteristics and Genetic Evolution of Echovirus 33 in Mainland of China. Pathogens. 2022; 11(11):1379. https://doi.org/10.3390/pathogens11111379
Chicago/Turabian StyleWang, Wenhui, Huan Fan, Shuaifeng Zhou, Shikang Li, Alitengsaier NIGEDELI, Yong Zhang, Qiang Sun, Yun He, Qin Guo, Xiaoyi Wang, and et al. 2022. "Molecular Characteristics and Genetic Evolution of Echovirus 33 in Mainland of China" Pathogens 11, no. 11: 1379. https://doi.org/10.3390/pathogens11111379
APA StyleWang, W., Fan, H., Zhou, S., Li, S., NIGEDELI, A., Zhang, Y., Sun, Q., He, Y., Guo, Q., Wang, X., Lu, H., Xiao, J., Zhao, H., Han, Z., Ji, T., Zhang, L., & Yan, D. (2022). Molecular Characteristics and Genetic Evolution of Echovirus 33 in Mainland of China. Pathogens, 11(11), 1379. https://doi.org/10.3390/pathogens11111379