
Proteomic Analysis of Leishmania donovani Membrane Components Reveals the Role of Activated Protein C Kinase in Host-Parasite Interaction


Abstract
1. Introduction
2. Results
2.1. Host-Parasite Interaction
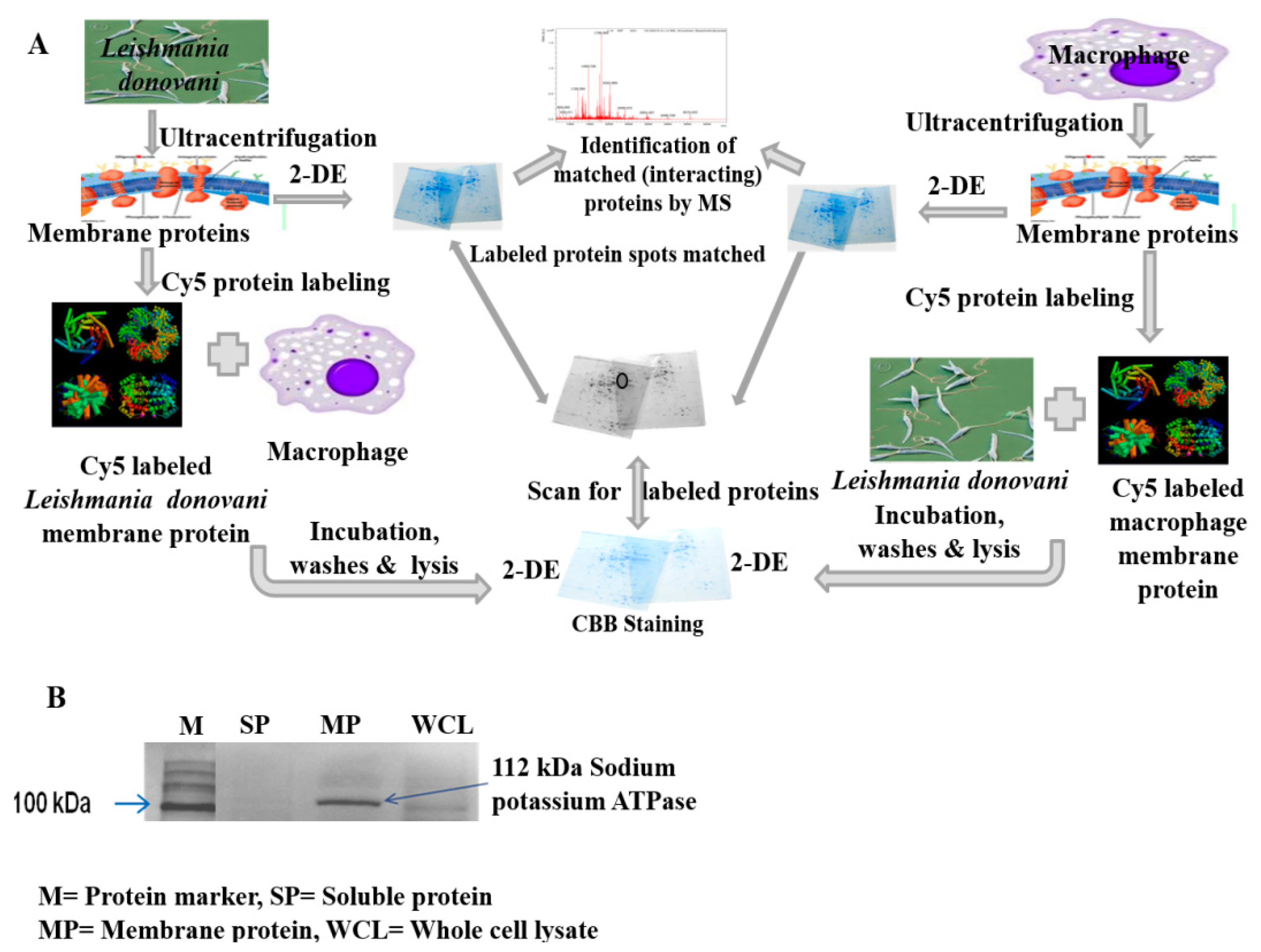
2.1.1. Interaction between Parasite and Macrophage Membrane Protein
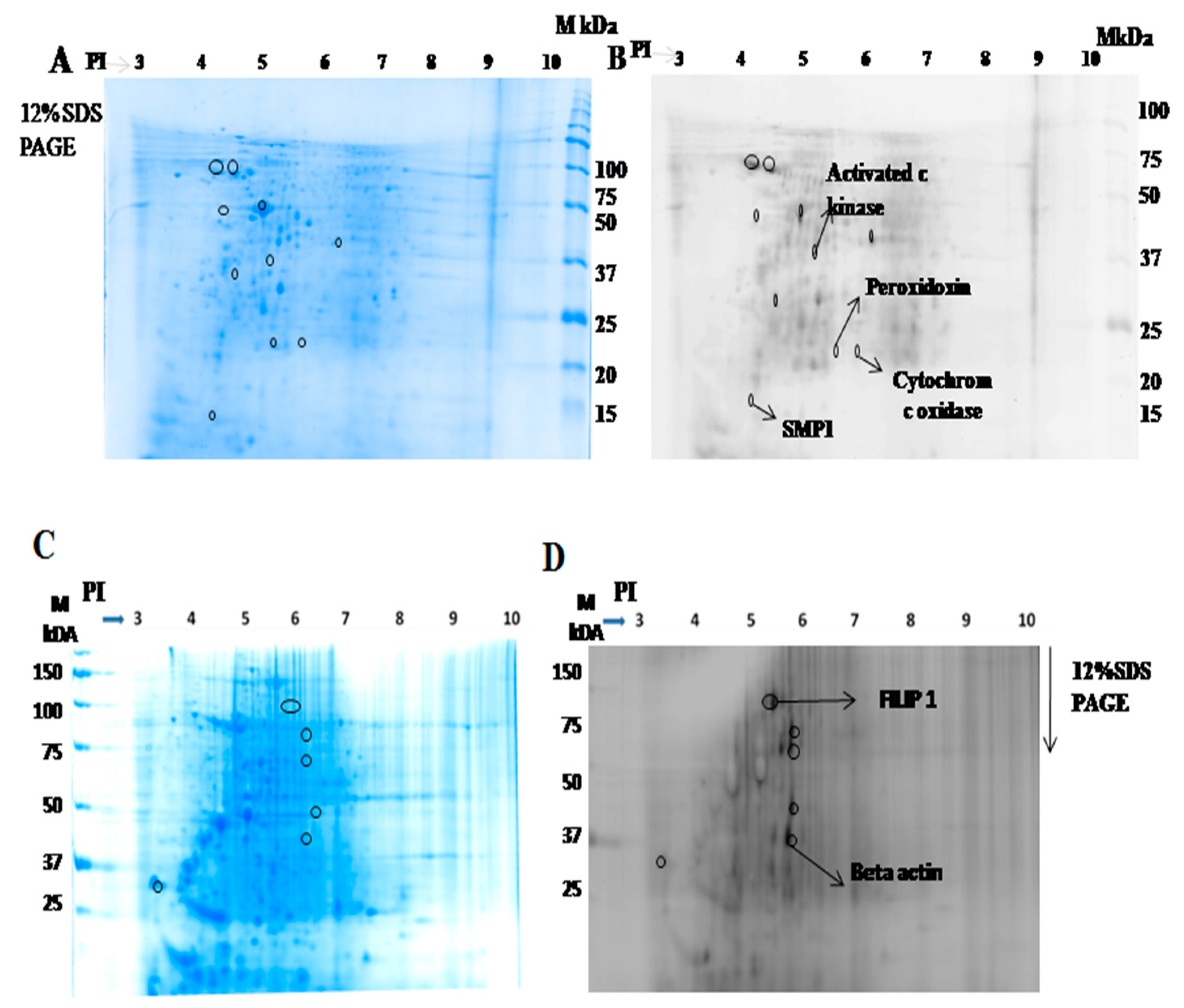
2.1.2. Identification of Interacting Membrane Proteins by 2-DE and MS
2.1.3. Cytotoxicity of Withaferin
2.1.4. Activated C kinase Protein Favors Parasite Proliferation within Host
3. Discussion
4. Materials and Methods
4.1. Parasite Culture
4.2. Macrophage Culture
4.3. Isolation and Quantification of Membrane Proteins
4.4. Verifying Integrity and Purity of Membrane Proteins
4.5. Evaluation of Protein Interaction by ELISA
4.6. Identification of Membrane Proteins Involved in Host-Parasite Interaction
4.7. Two-Dimensional Gel Electrophoresis (2 DE)
Iso-Electric Focusing (IEF)
4.8. SDS-PAGE
4.9. In-Gel Digestion
4.10. Protein Identification by MALDI-TOF/TOF
4.11. Sensitivity of Leishmania Promastigote towards Withaferin A
4.12. Cytotoxicity of WA on THP-1 Macrophages
4.13. Proliferation of L. donovani Parasite within Host Macrophage in Presence and Absence of Activated C kinase Inhibitor (Withaferin A)
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- LaMotte, S.; Späth, G.F.; Rachidi, N.; Prina, E. The enemy within: Targeting host–parasite interaction for antileishmanial drug discovery. PLoS Negl. Trop. Dis. 2017, 11, e0005480. [Google Scholar] [CrossRef] [PubMed]
- Sundar, S.; Singh, B. Understanding Leishmania parasites through proteomics and implications for the clinic. Expert Rev. Proteom. 2018, 15, 371–390. [Google Scholar] [CrossRef] [PubMed]
- Cantacessi, C.; Dantas-Torres, F.; Nolan, M.J.; Otranto, D. The past, present, and future of Leishmania genomics and transcriptomics. Trends Parasitol. 2015, 31, 100–108. [Google Scholar] [CrossRef]
- Brobey, R.K.B.; Mei, F.C.; Cheng, X.; Soong, L. Comparative two-dimensional gel electrophoresis maps for promastigotes of Leishmania amazonensis and Leishmania major. Braz. J. Infect. Dis. 2006, 10, 1–6. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lynn, M.A.; Marr, A.; McMaster, W.R. Differential quantitative proteomic profiling of Leishmania infantum and Leishmania mexicana density gradient separated membranous fractions. J. Proteom. 2013, 82, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Capelli-Peixoto, J.; Mule, S.N.; Tano, F.T.; Palmisano, G.; Stolf, B.S. Proteomics and Leishmaniasis: Potential Clinical Applications. Proteom. Clin. Appl. 2019, 13, e1800136. [Google Scholar] [CrossRef]
- Sundar, S.; Singh, B. Identifying vaccine targets for anti-leishmanial vaccine development. Expert Rev. Vaccines 2014, 13, 489–505. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Uzonna, J.E. The early interaction of Leishmania with macrophages and dendritic cells and its influence on the host immune response. Front. Cell. Infect. Microbiol. 2012, 12, 83. [Google Scholar] [CrossRef] [PubMed]
- Kedzierski, L.; Montgomery, J.; Bullen, D.; Curtis, J.; Gardiner, E.; Jimenez-Ruiz, A.; Handman, E. A Leucine-Rich Repeat Motif of Leishmania Parasite Surface Antigen 2 Binds to Macrophages through the Complement Receptor 3. J. Immunol. 2004, 172, 4902–4906. [Google Scholar] [CrossRef]
- Alexander, J.; Satoskar, A.R.; Russell, D.G. Leishmania species: Models of intracellular parasitism. J. Cell Sci. 1999, 112, 2993–3002. [Google Scholar] [CrossRef]
- McMahon-Pratt, D.; Alexander, J. Does the Leishmania major paradigm of pathogenesis and protection hold for New World cutaneous leishmaniases or the visceral disease? Immunol. Rev. 2004, 201, 206–224. [Google Scholar] [CrossRef]
- Young, J.; Kima, P.E. The Leishmania Parasitophorous Vacuole Membrane at the Parasite-Host Interface. Yale J. Biol. Med 2019, 92, 511–521. [Google Scholar]
- Silverman, J.M.; Clos, J.; De’Oliveira, C.C.; Shirvani, O.; Fang, Y.; Wang, C.; Foster, L.J.; Reiner, N.E. An exosome-based secretion pathway is responsible for protein export from Leishmania and communication with macrophages. J. Cell Sci. 2010, 123, 842–852. [Google Scholar] [CrossRef] [PubMed]
- Matlashewski, G. Leishmania infection and virulence. Med. Microbiol. Immunol. 2001, 190, 37–42. [Google Scholar] [CrossRef]
- Garg, G.; Ali, V.; Singh, K.; Gupta, P.; Ganguly, A.; Sahasrabuddhe, A.A.; Das, P. Quantitative secretome analysis unravels new secreted proteins in Amphotericin B resistant Leishmania donovani. J. Proteom. 2019, 207, 103464. [Google Scholar] [CrossRef]
- Podinovskaia, M.; Descoteaux, A. Leishmania and the macrophage: A multifaceted interaction. Futur. Microbiol. 2015, 10, 111–129. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Flohé, L. Is there a role of glutathione peroxidases in signaling and differentiation? BioFactors 2003, 17, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Rocha, M.C.; De Godoy, K.F.; Bannitz-Fernandes, R.; Fabri, J.H.T.M.; Barbosa, M.M.F.; De Castro, P.A.; Almeida, F.; Goldman, G.H.; Da Cunha, A.F.; Netto, L.E.S.; et al. Analyses of the three 1-Cys Peroxiredoxins from Aspergillus fumigatus reveal that cytosolic Prx1 is central to H2O2 metabolism and virulence. Sci. Rep. 2018, 8, 1–18. [Google Scholar] [CrossRef]
- Mandal, A.; Das, S.; Kumar, A.; Roy, S.; Verma, S.; Ghosh, A.K.; Singh, R.; Kumar, A.; Sainia, S.; Sardar, A.H.; et al. l-Arginine uptake by cationic amino acid transporter promotes intra-macrophage survival of Leishmania donovani by enhancing arginase-mediated polyamine synthesis. Front. Immunol. 2017, 8, 839. [Google Scholar] [CrossRef] [PubMed]
- Goel, R.; Murthy, K.R.; Srikanth, S.M.; Pinto, S.M.; Bhattacharjee, M.; Kelkar, D.S.; Madugundu, A.K.; Dey, G.; Mohan, S.S.; Krishna, V.; et al. Characterizing the normal proteome of human ciliary body. Clin. Proteom. 2013, 10, 9. [Google Scholar] [CrossRef]
- Kumar, A.; Misra, P.; Sisodia, B.; Shasany, A.K.; Sundar, S.; Dube, A. Proteomic analyses of membrane enriched proteins of Leishmania donovani Indian clinical isolate by mass spectrometry. Parasitol. Int. 2015, 64, 36–42. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Morales, M.A.; Watanabe, R.; Laurent, C.; Lenormand, P.; Rousselle, J.-C.; Namane, A.; Späth, G.F. Phosphoproteomic analysis of Leishmania donovani pro- and amastigote stages. Proteomics 2008, 8, 350–363. [Google Scholar] [CrossRef]
- Gretes, M.C.; Poole, L.B.; Karplus, P.A. Peroxiredoxins in Parasites. Antioxid. Redox Signal. 2012, 17, 608–633. [Google Scholar] [CrossRef]
- Tull, D.; Vince, J.E.; Callaghan, J.M.; Naderer, T.; Spurck, T.; McFadden, G.I.; Currie, G.; Ferguson, K.; Bacic, A.; McConville, M.J. SMP-1, a Member of a New Family of Small Myristoylated Proteins in Kinetoplastid Parasites, Is Targeted to the Flagellum Membrane in Leishmania. Mol. Biol. Cell 2004, 15, 4775–4786. [Google Scholar] [CrossRef]
- Taylor, C.M.; Fischer, K.; Abubucker, S.; Wang, Z.; Martin, J.; Jiang, D.; Magliano, M.; Rosso, M.-N.; Li, B.-W.; Fischer, P.U.; et al. Targeting Protein-Protein Interactions for Parasite Control. PLoS ONE 2011, 6, e18381. [Google Scholar] [CrossRef]
- Afrin, F.; Khan, I.; Hemeg, H.A. Leishmania-Host Interactions—An Epigenetic Paradigm. Front. Immunol. 2019, 10, 492. [Google Scholar] [CrossRef] [PubMed]
- Shio, M.T.; Hassani, K.; Isnard, A.; Ralph, B.; Contreras, I.; Gomez, M.A.; Abu-Dayyeh, I.; Olivier, M. Host Cell Signalling and Leishmania: Mechanisms of Evasion. J. Trop. Med. 2012, 2012, 1–14. [Google Scholar] [CrossRef]
- Kelly, B.L.; Stetson, D.B.; Locksley, R.M. Leishmania major LACK Antigen Is Required for Efficient Vertebrate Parasitization. J. Exp. Med. 2003, 198, 1689–1698. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, D.; Carter, P.M.; Nation, C.S.; Pizarro, J.C.; Guidry, J.; Aiyar, A.; Kelly, B.L. LACK, a RACK1 ortholog, facilitates cytochrome c oxidase subunit expression to promote Leishmania major fitness. Mol. Microbiol. 2015, 96, 95–109. [Google Scholar] [CrossRef]
- Sharma, U.; Velpandian, T.; Sharma, P.; Singh, S.; Singh, S. Evaluation of anti-leishmanial activity of selected Indian plants known to have antimicrobial properties. Parasitol. Res. 2009, 105, 1287–1293. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, S.; Veronica, J.; Gundampati, R.K.; Sundar, S.; Maurya, R. Exploring the inhibitory activity of Withaferin-A against Pteridine reductase-1 of L. donovani. J. Enzym. Inhib. Med. Chem. 2015, 31, 1029–1037. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Roy, S.; Kumar, G.A.; Jafurulla, M.; Mandal, C.; Chattopadhyay, A. Integrity of the Actin Cytoskeleton of Host Macrophages is Essential for Leishmania donovani Infection. Biochim. Biophys. Acta 2014, 1838, 2011–2018. [Google Scholar] [CrossRef]
- Yue, J.; Huhn, S.; Shen, Z. Complex roles of filamin-A mediated cytoskeleton network in cancer progression. Cell Biosci. 2013, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Kulshrestha, A.; Bhandari, V.; Mukhopadhyay, R.; Ramesh, V.; Sundar, S.; Maes, L.; Dujardin, J.C.; Roy, S.; Salotra, P. Validation of a simple resazurin-based promastigote assay for the routine monitoring of miltefosine susceptibility in clinical isolates of Leishmania donovani. Parasitol. Res. 2013, 112, 825–828. [Google Scholar] [CrossRef]
- Kumar, D.; Kulshrestha, A.; Singh, R.; Salotra, P. In Vitro Susceptibility of Field Isolates of Leishmania donovani to Miltefosine and Amphotericin B: Correlation with Sodium Antimony Gluconate Susceptibility and Implications for Treatment in Areas of Endemicity. Antimicrob. Agents Chemother. 2008, 53, 835–838. [Google Scholar] [CrossRef]
- Da Luz, R.I.; Vermeersch, M.; Dujardin, J.-C.; Cos, P.; Maes, L. In Vitro Sensitivity Testing of Leishmania Clinical Field Isolates: Preconditioning of Promastigotes Enhances Infectivity for Macrophage Host Cells. Antimicrob. Agents Chemother. 2009, 53, 5197–5203. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nesterenko, M.V.; Woods, K.; Upton, S.J. Receptor/ligand interactions between Cryptosporidium parvum and the surface of the host cell. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 1999, 1454, 165–173. [Google Scholar] [CrossRef][Green Version]
- Foryst-Ludwig, A.; Kreissl, M.C.; Benz, V.; Brix, S.; Smeir, E.; Ban, Z.; Januszewicz, E.; Salatzki, J.; Grune, J.; Schwanstecher, A.-K.; et al. Adipose Tissue Lipolysis Promotes Exercise-induced Cardiac Hypertrophy Involving the Lipokine C16:1n7-Palmitoleate. J. Biol. Chem. 2015, 290, 23603–23615. [Google Scholar] [CrossRef]
- Ocaña-Fuentes, A.; Gutiérrez, E.A.; Señorans, F.; Reglero, G. Supercritical fluid extraction of oregano (Origanum vulgare) essentials oils: Anti-inflammatory properties based on cytokine response on THP-1 macrophages. Food Chem. Toxicol. 2010, 48, 1568–1575. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Plate Coating | Interaction | Primary Antibody | Mean OD ± SD |
|---|---|---|---|
| Blank (BSA only) | Parasite MP | VL patient serum | 0.0 |
| THP-1 MP | Parasite MP | VL patient serum | 0.96375 ± 0.03 * |
| THP-1 Lysate | Parasite MP | VL patient serum | 1.07755 ± 0.02 * |
| THP-1 Lysate | None | VL patient serum | 0.50365 ± 0.02 * |
| Parasite MP | None | VL patient serum | 1.29775 ± 0.04 |
| S.No | Protein Name | UniProt ID | Peptide Sequence |
|---|---|---|---|
| 1 | Activated protein c kinase | E9BK16 | K.INVESPINQIAFSPNR.F |
| 2 | Peroxidoxin | E9BG25 | R.HSTINDLPVGR.N |
| 3 | Cytochrome c oxidase | E9AGF4 | R.WNLNTELHPADR.A |
| 4 | SMP 1 | A4HYX4 | K.MDALPLSEEYR.Q |
| 5 | FILIP 1 | Q7Z7B0 | K.SSELSCSVDLLK.K |
| 6 | Beta actin | Q53G99 | K.DLYANTVLSGGTTMYPGIADR.M |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verma, S.; Deep, D.K.; Gautam, P.; Singh, R.; Salotra, P. Proteomic Analysis of Leishmania donovani Membrane Components Reveals the Role of Activated Protein C Kinase in Host-Parasite Interaction. Pathogens 2021, 10, 1194. https://doi.org/10.3390/pathogens10091194
Verma S, Deep DK, Gautam P, Singh R, Salotra P. Proteomic Analysis of Leishmania donovani Membrane Components Reveals the Role of Activated Protein C Kinase in Host-Parasite Interaction. Pathogens. 2021; 10(9):1194. https://doi.org/10.3390/pathogens10091194
Chicago/Turabian StyleVerma, Sandeep, Deepak Kumar Deep, Poonam Gautam, Ruchi Singh, and Poonam Salotra. 2021. "Proteomic Analysis of Leishmania donovani Membrane Components Reveals the Role of Activated Protein C Kinase in Host-Parasite Interaction" Pathogens 10, no. 9: 1194. https://doi.org/10.3390/pathogens10091194
APA StyleVerma, S., Deep, D. K., Gautam, P., Singh, R., & Salotra, P. (2021). Proteomic Analysis of Leishmania donovani Membrane Components Reveals the Role of Activated Protein C Kinase in Host-Parasite Interaction. Pathogens, 10(9), 1194. https://doi.org/10.3390/pathogens10091194