
The Distribution of Puumala orthohantavirus Genome Variants Correlates with the Regional Landscapes in the Trans-Kama Area of the Republic of Tatarstan

 ,
,  ,
, 
 and
and
Abstract
:1. Introduction
2. Materials and Methods
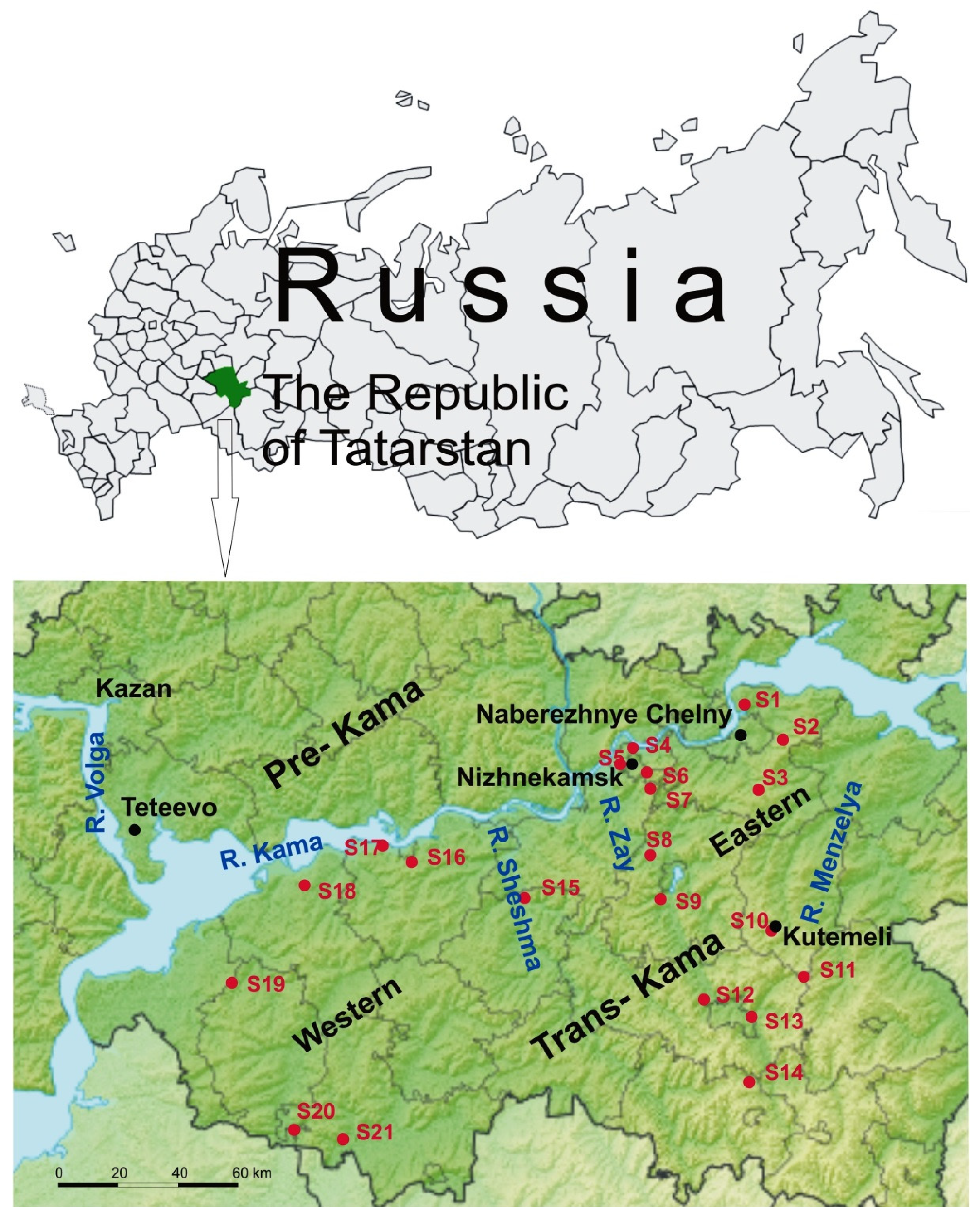
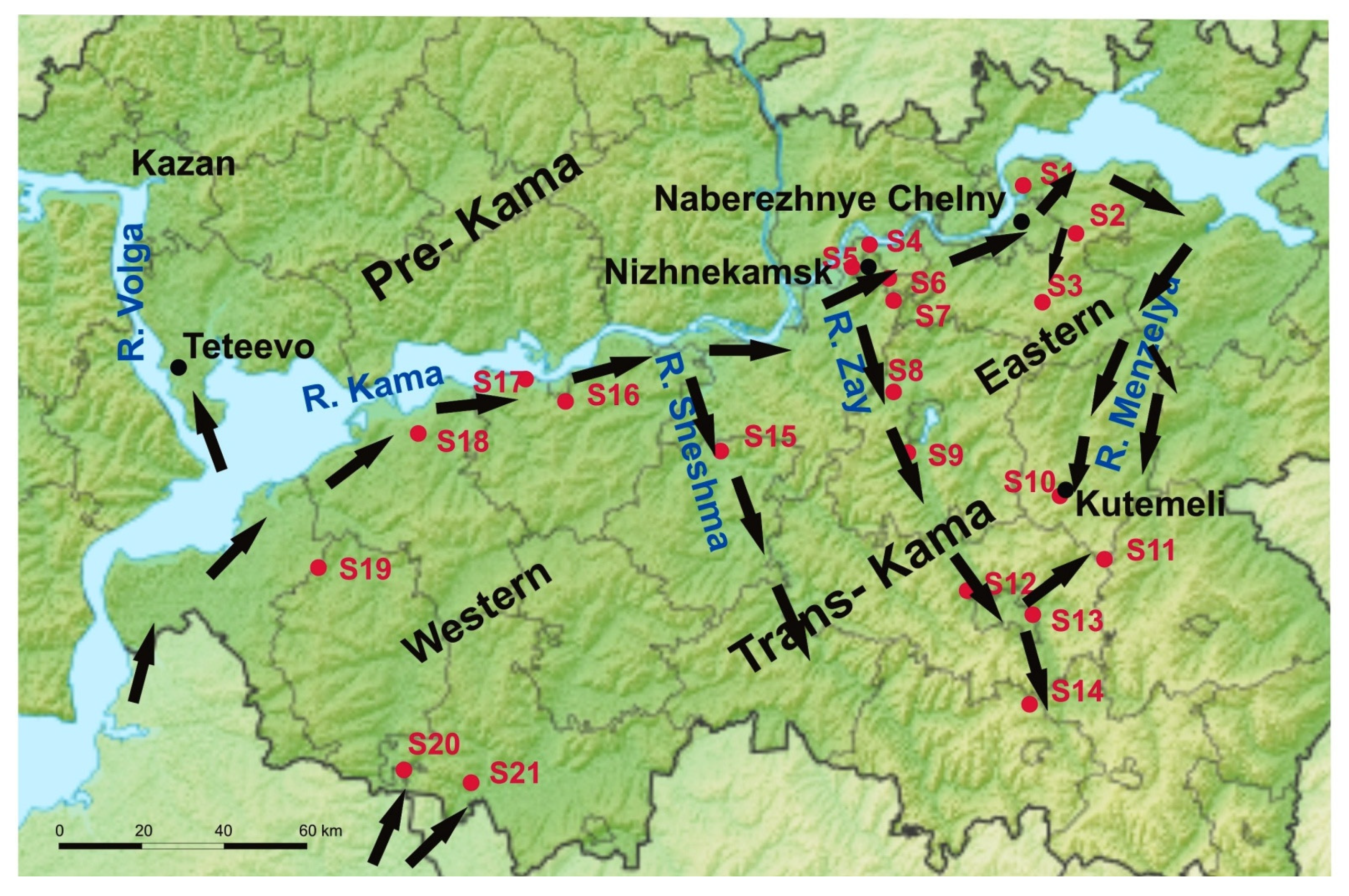
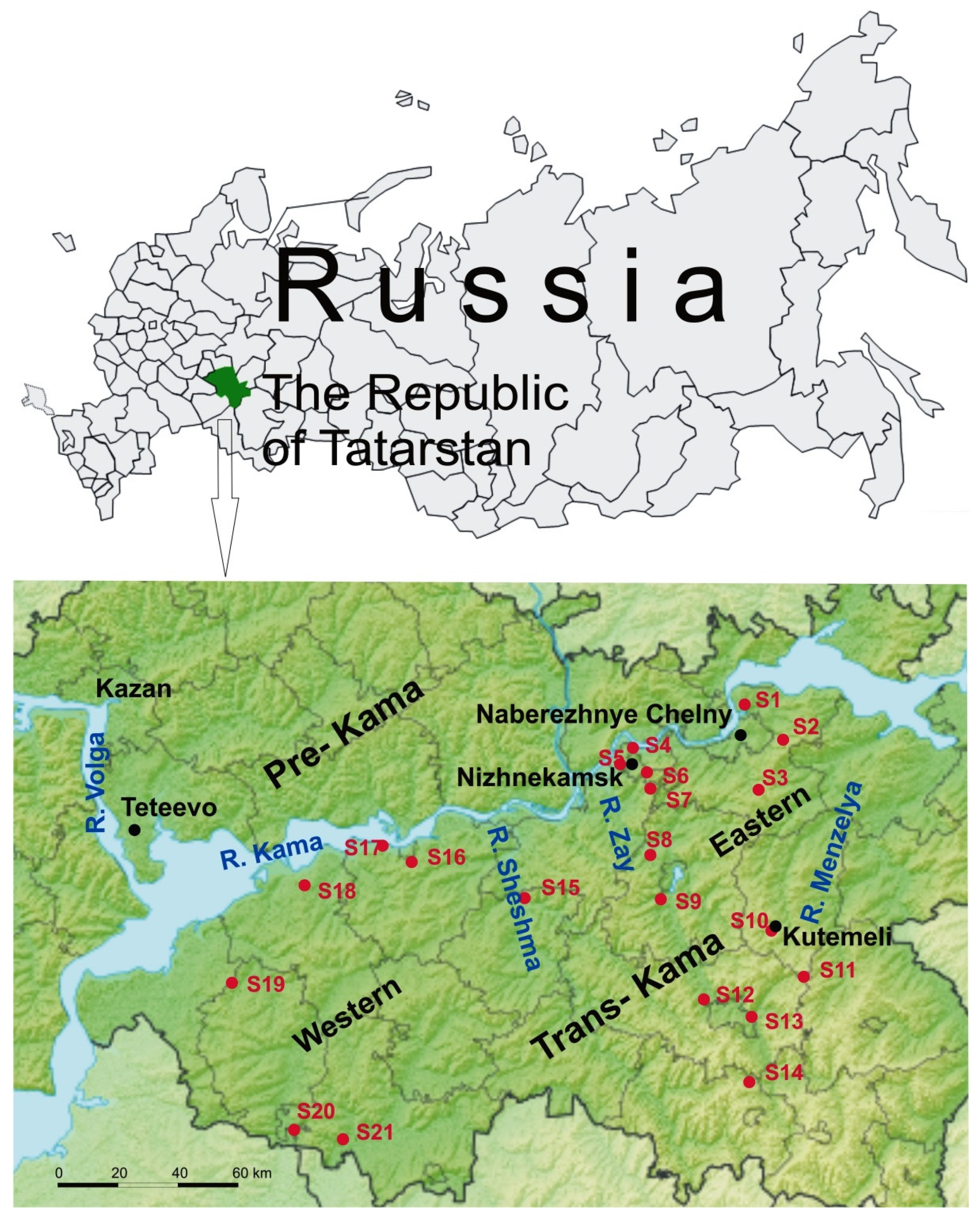
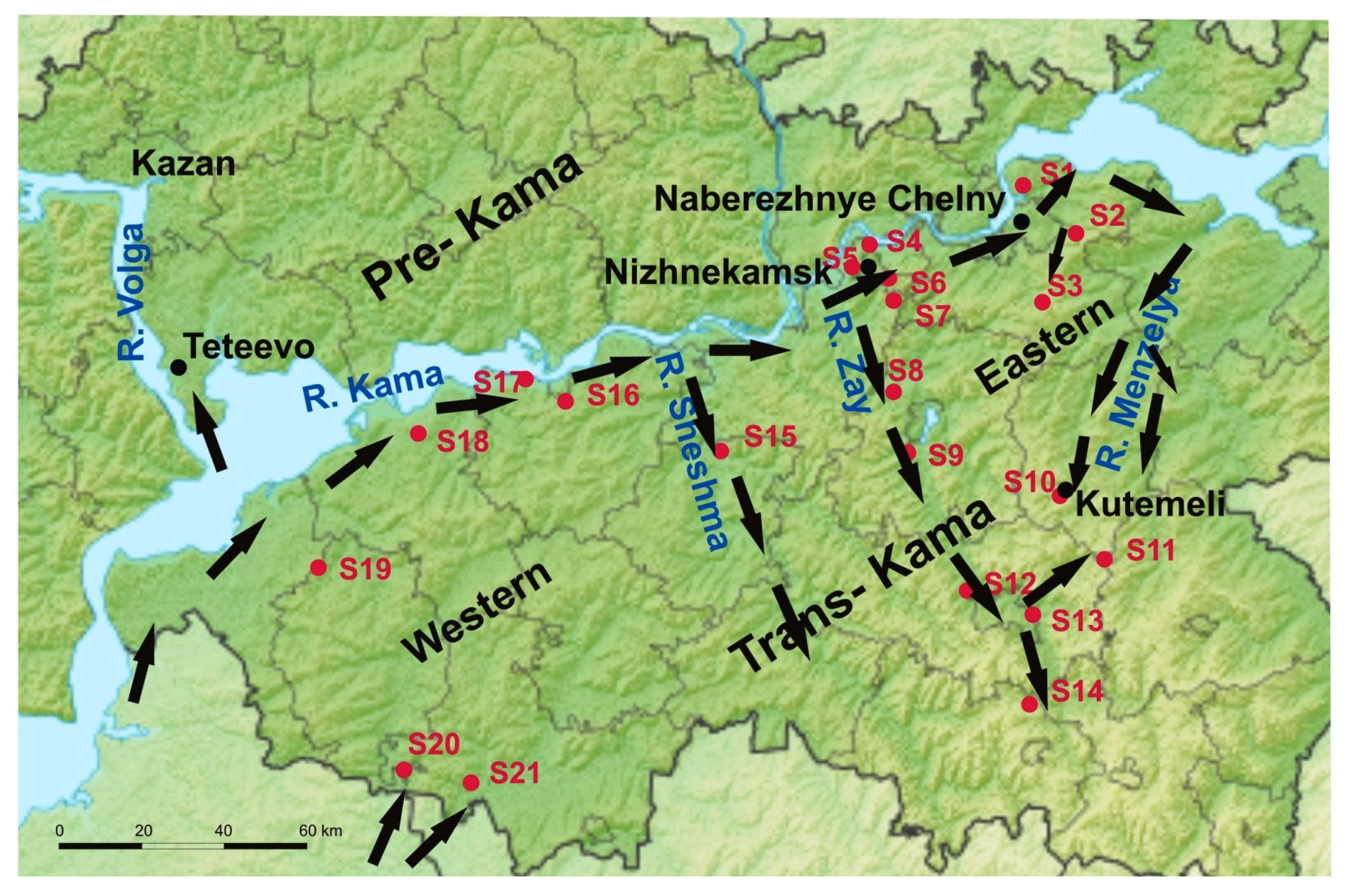
2.1. Rodent Tissues Sampling
2.2. RNA Extraction, cDNA Synthesis, and Polymerase Chain Reaction (PCR)
2.3. Phylogenetic Analysis
3. Results
3.1. Screening of Rodents
3.2. Molecular Analysis of the Trans-Kama PUUV Strains
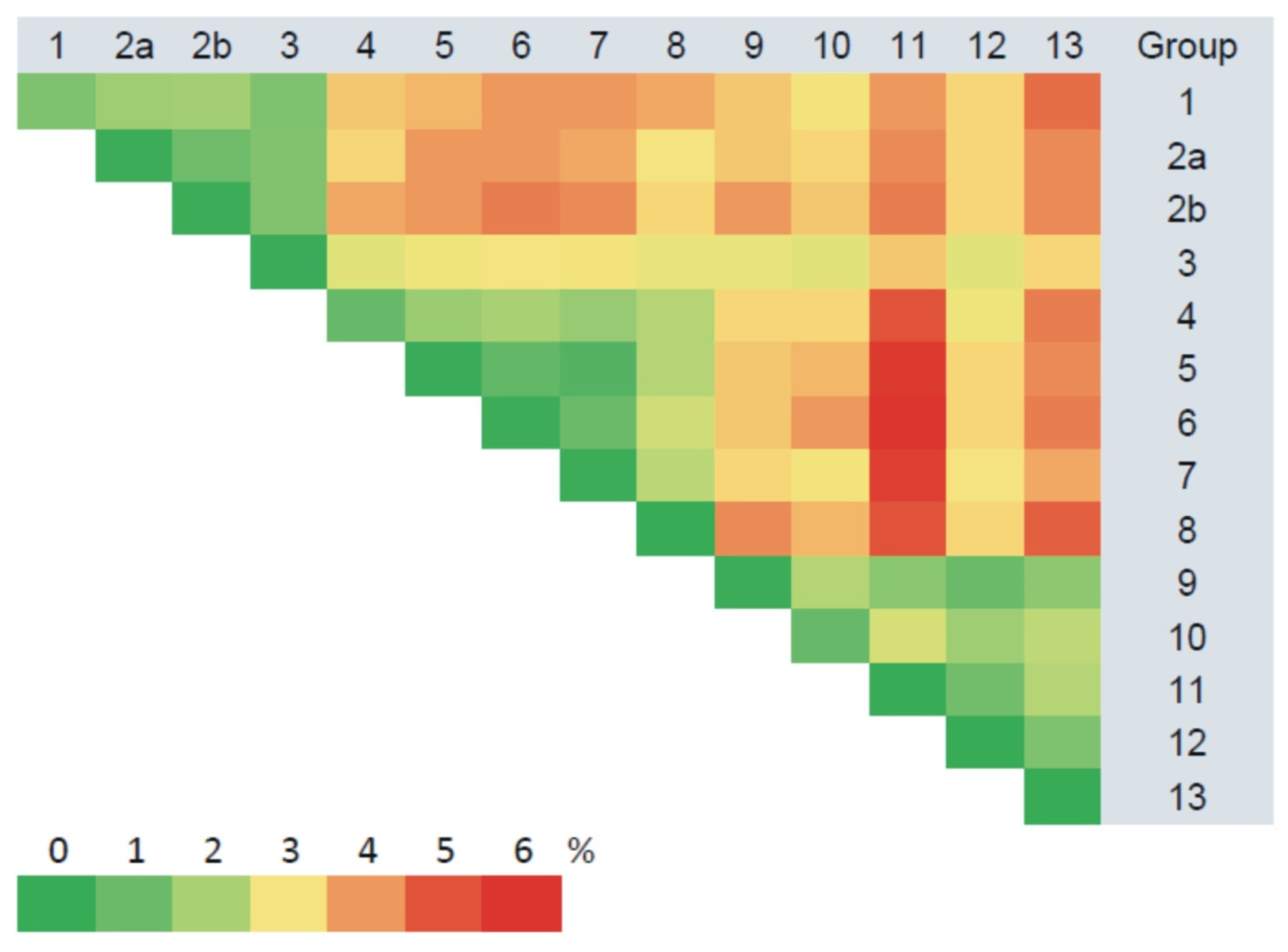
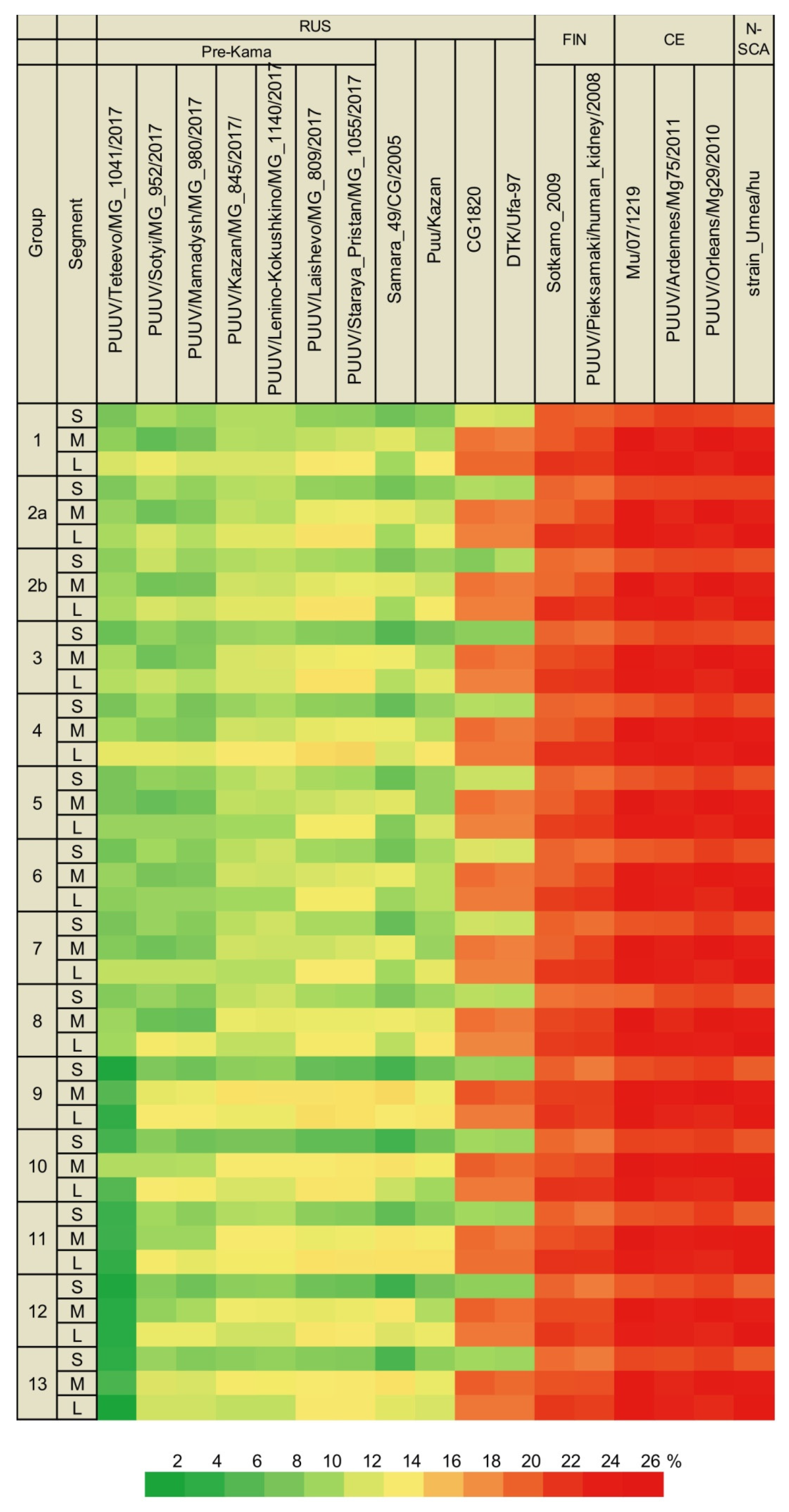
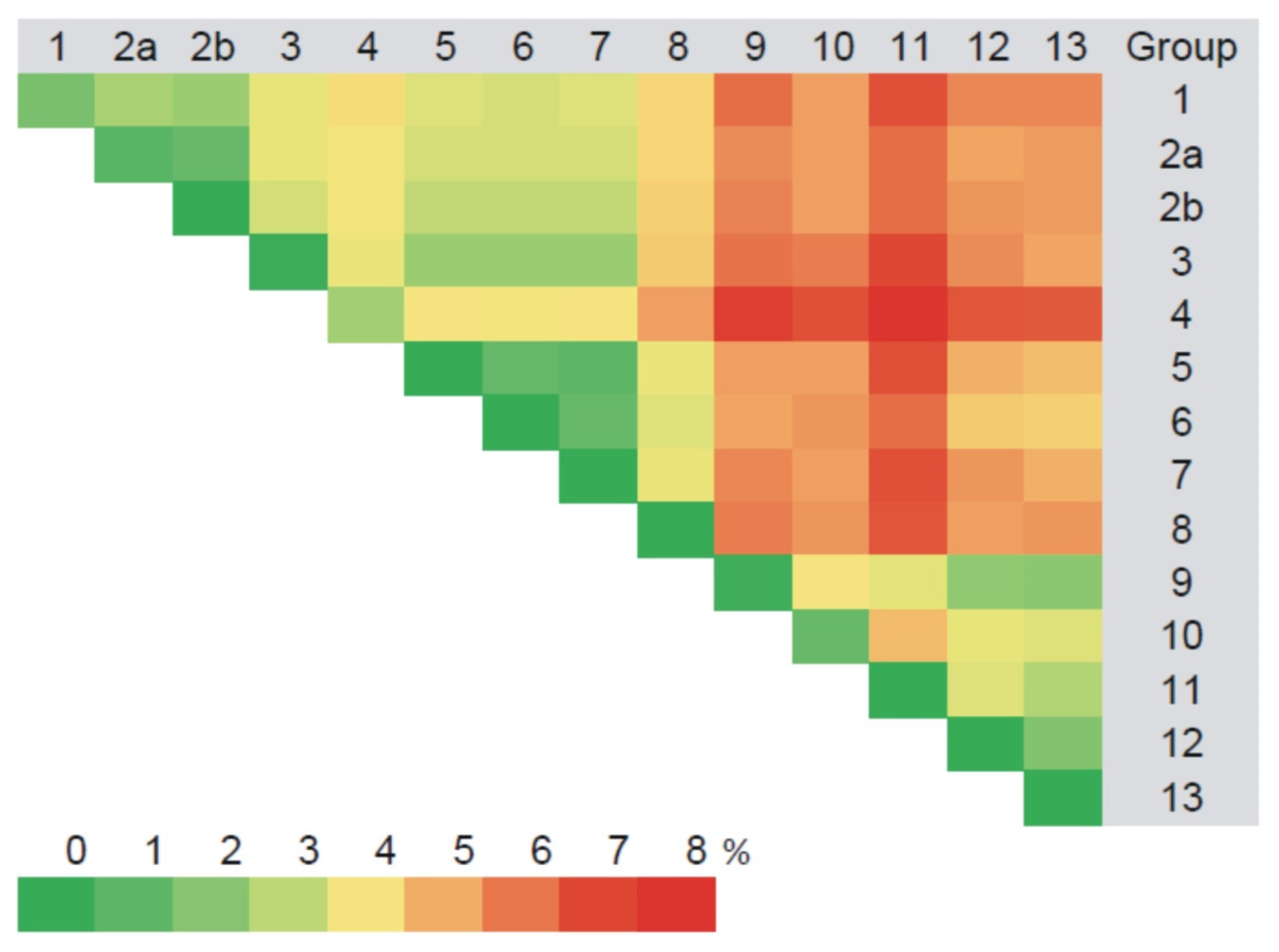
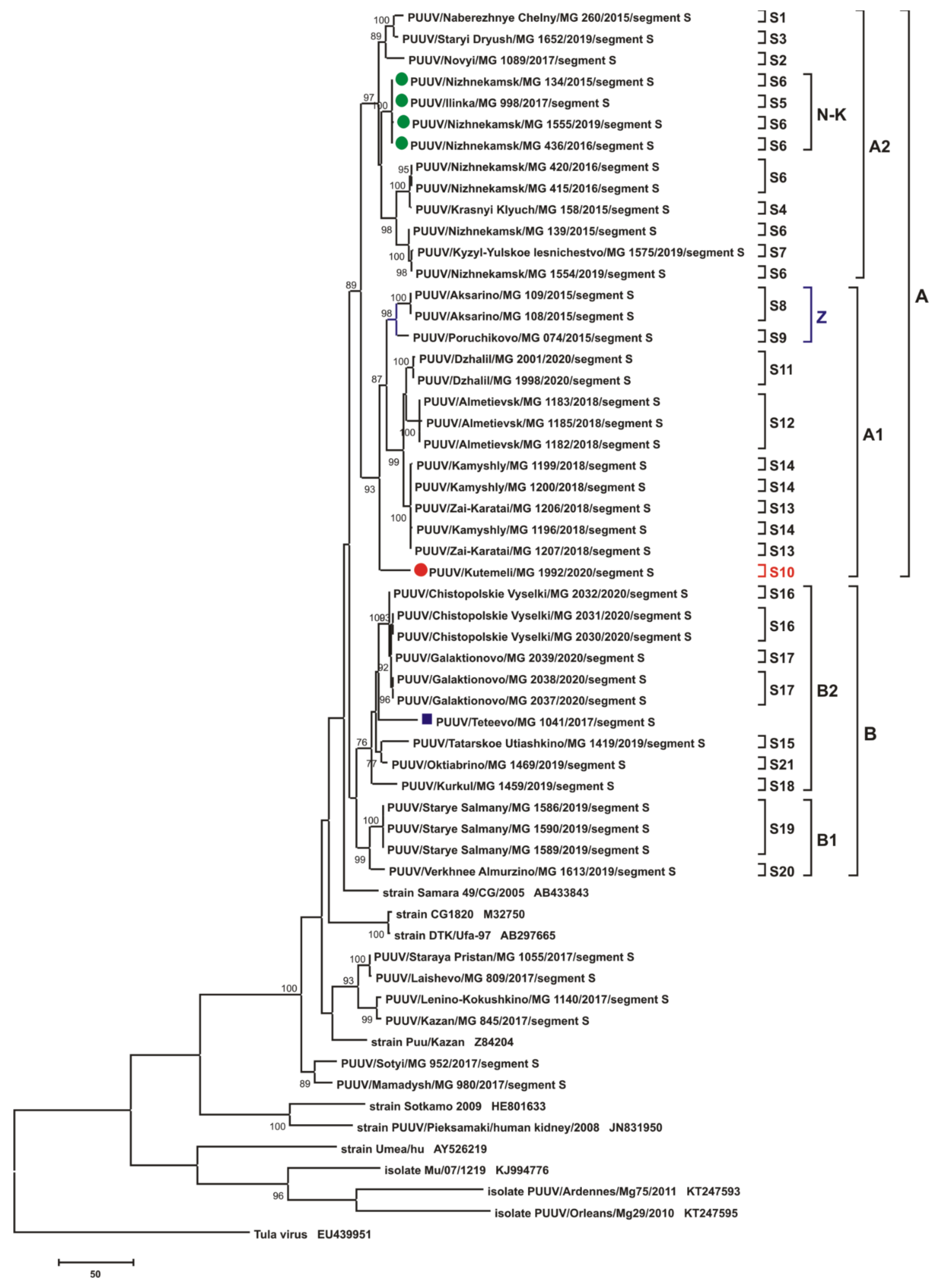
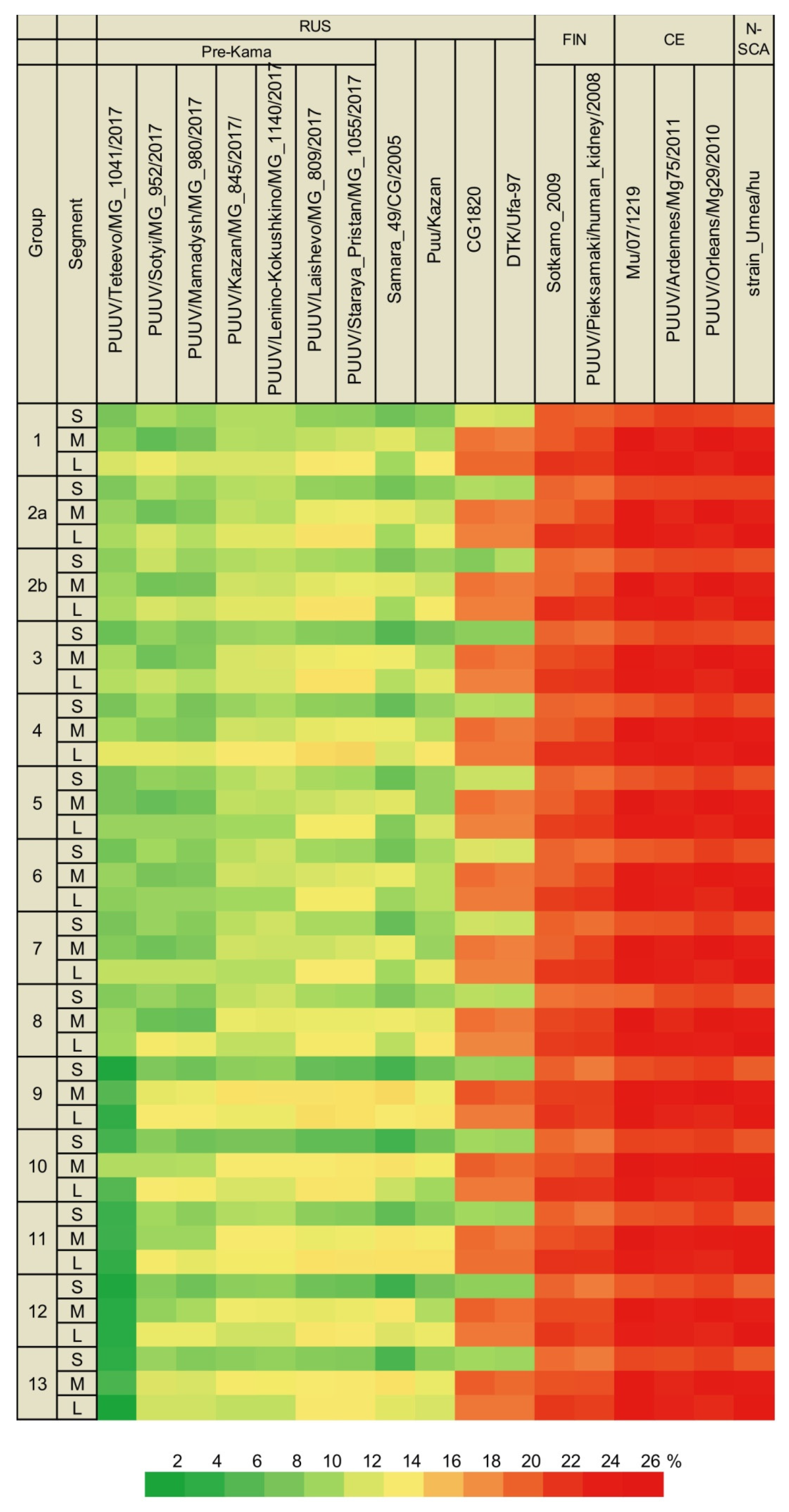
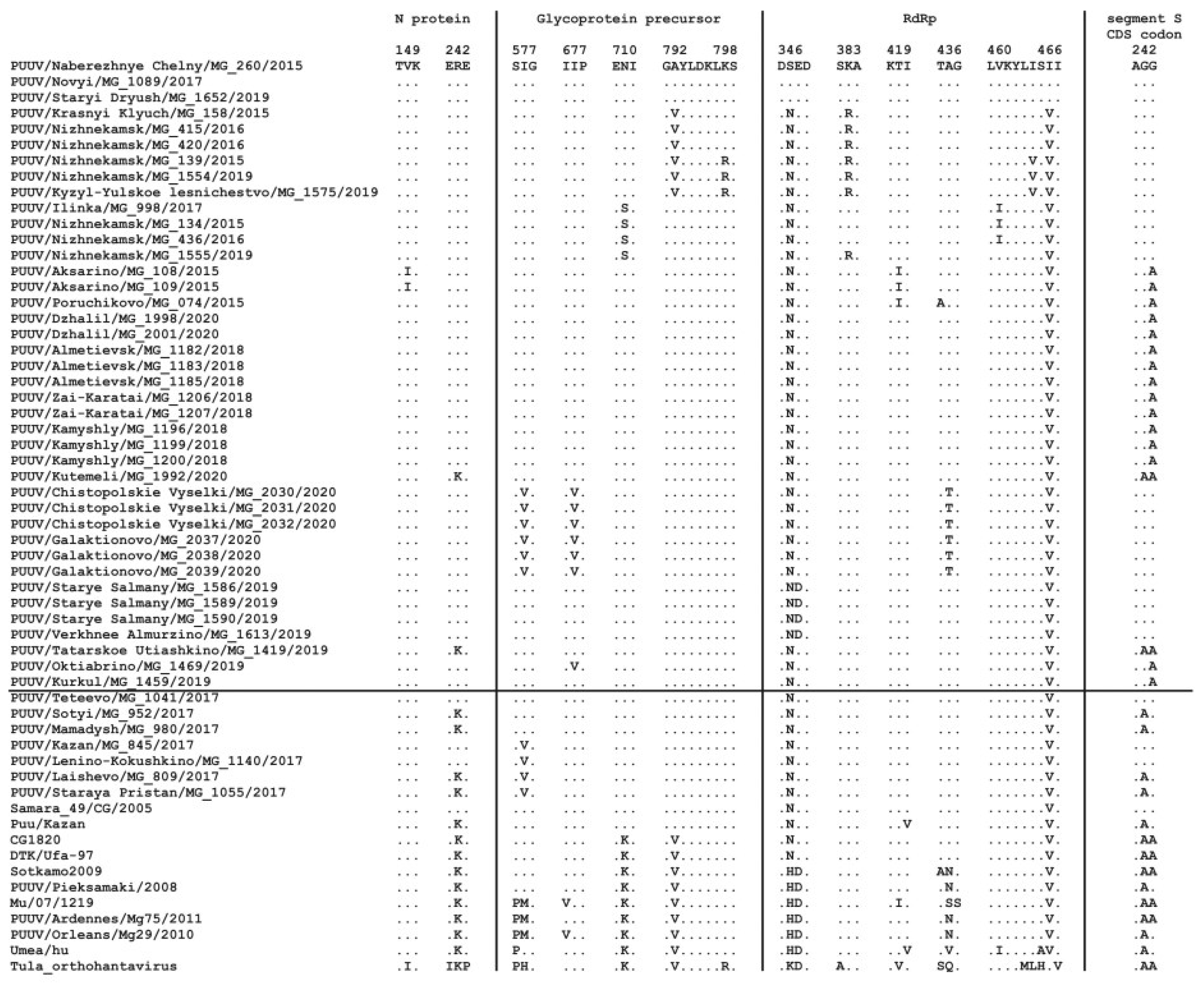
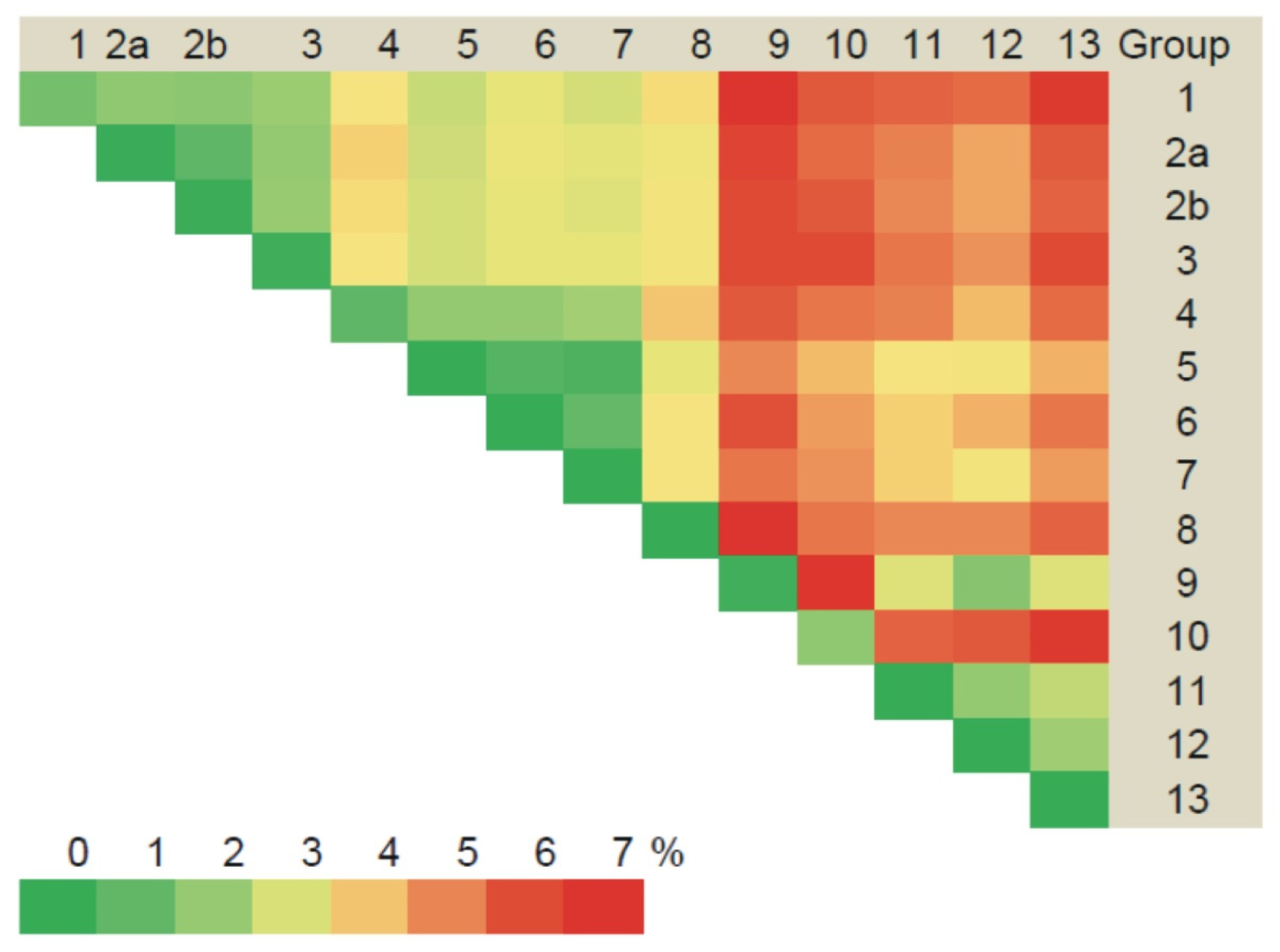
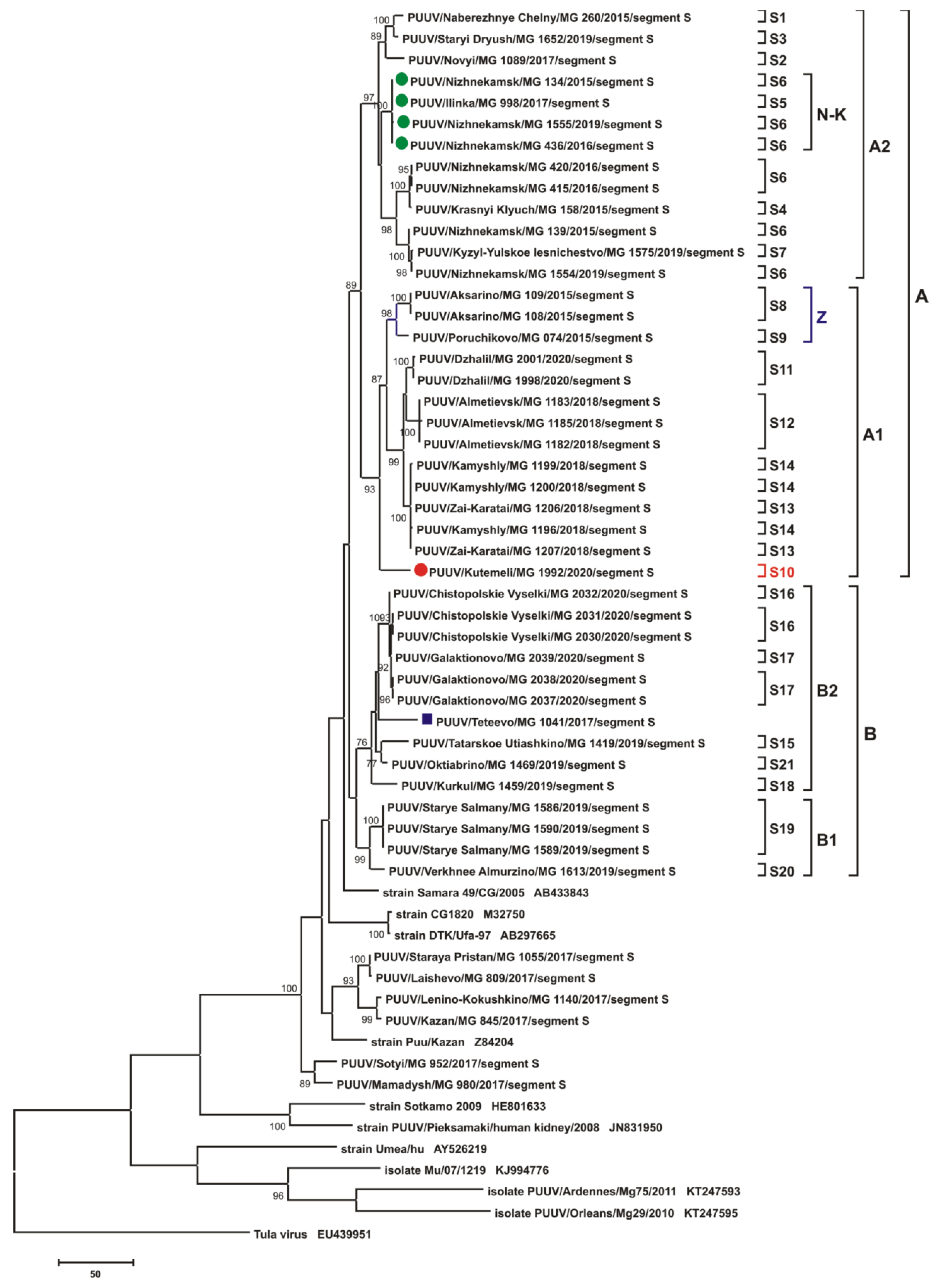
3.2.1. Comparison of the S Segment Nucleotide and N Protein Amino Acid Sequences of PUUV
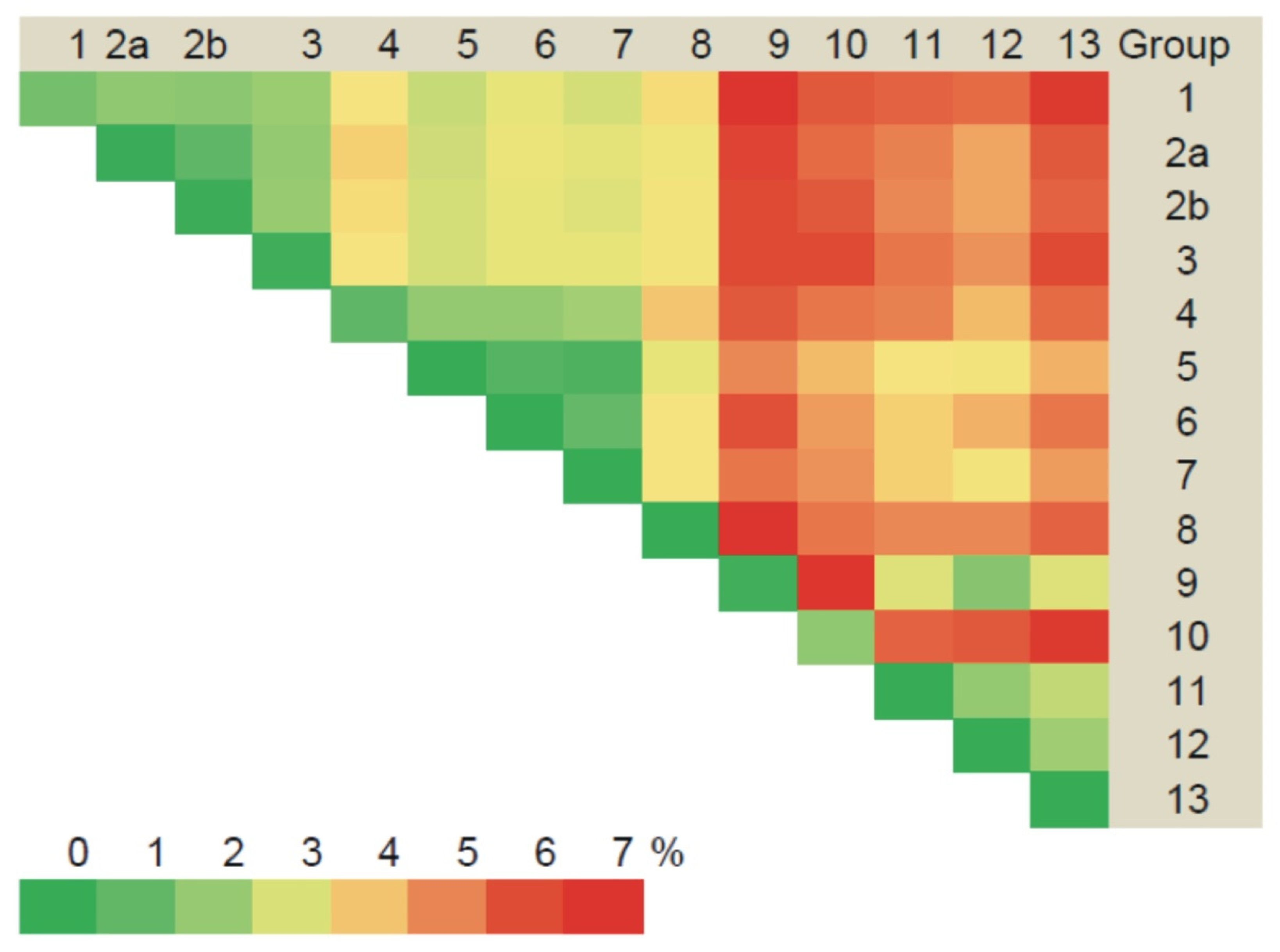
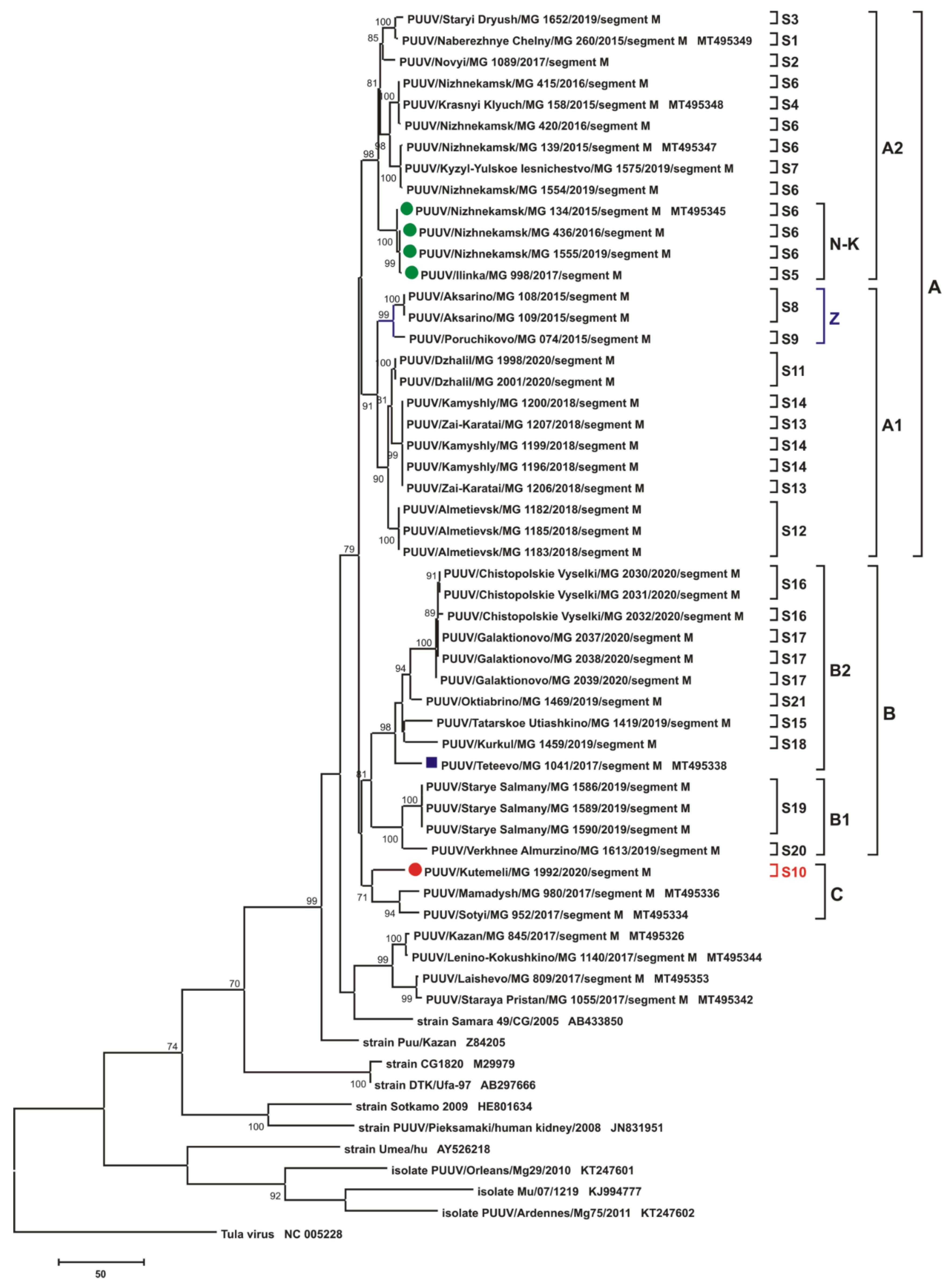
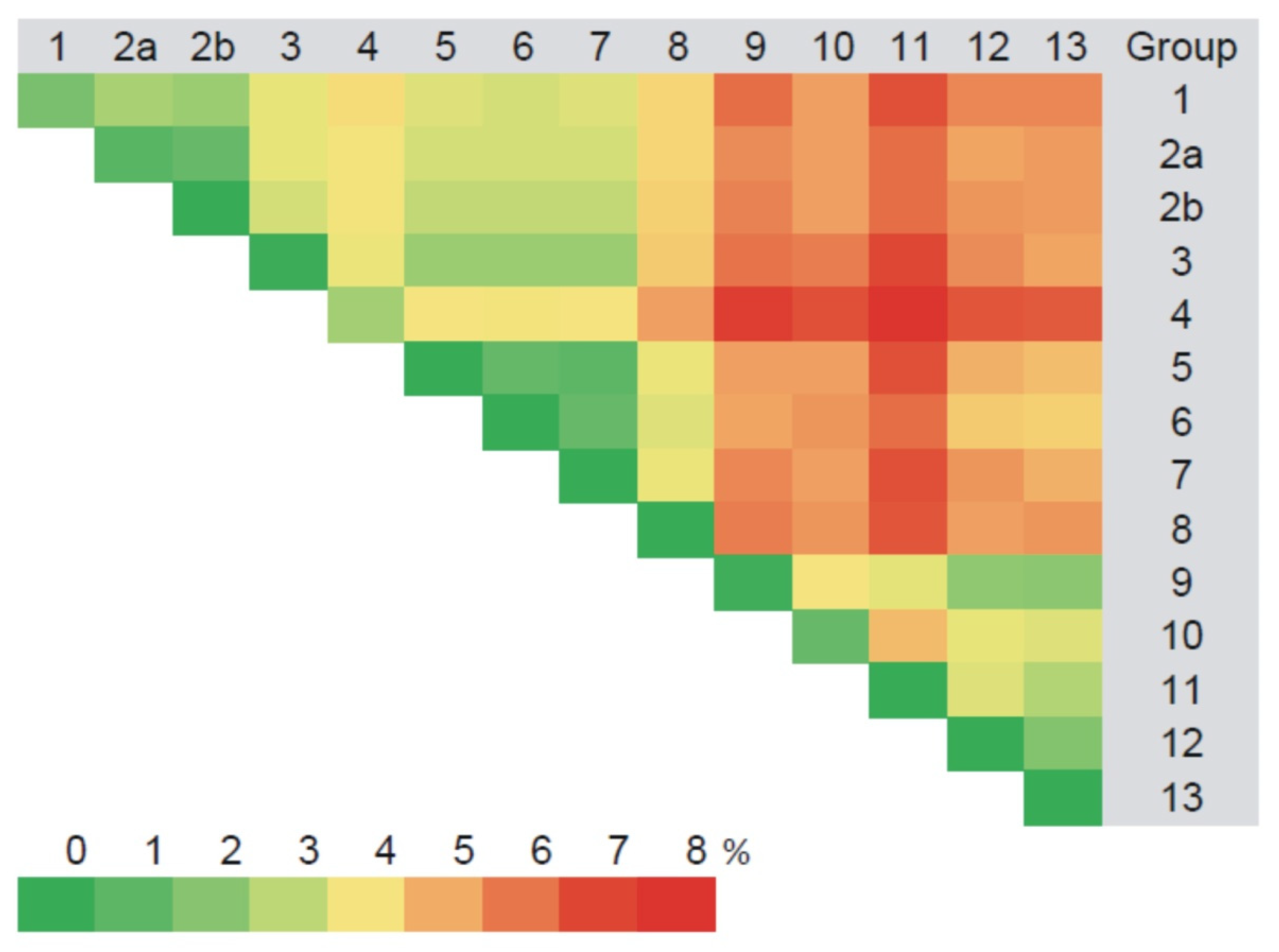
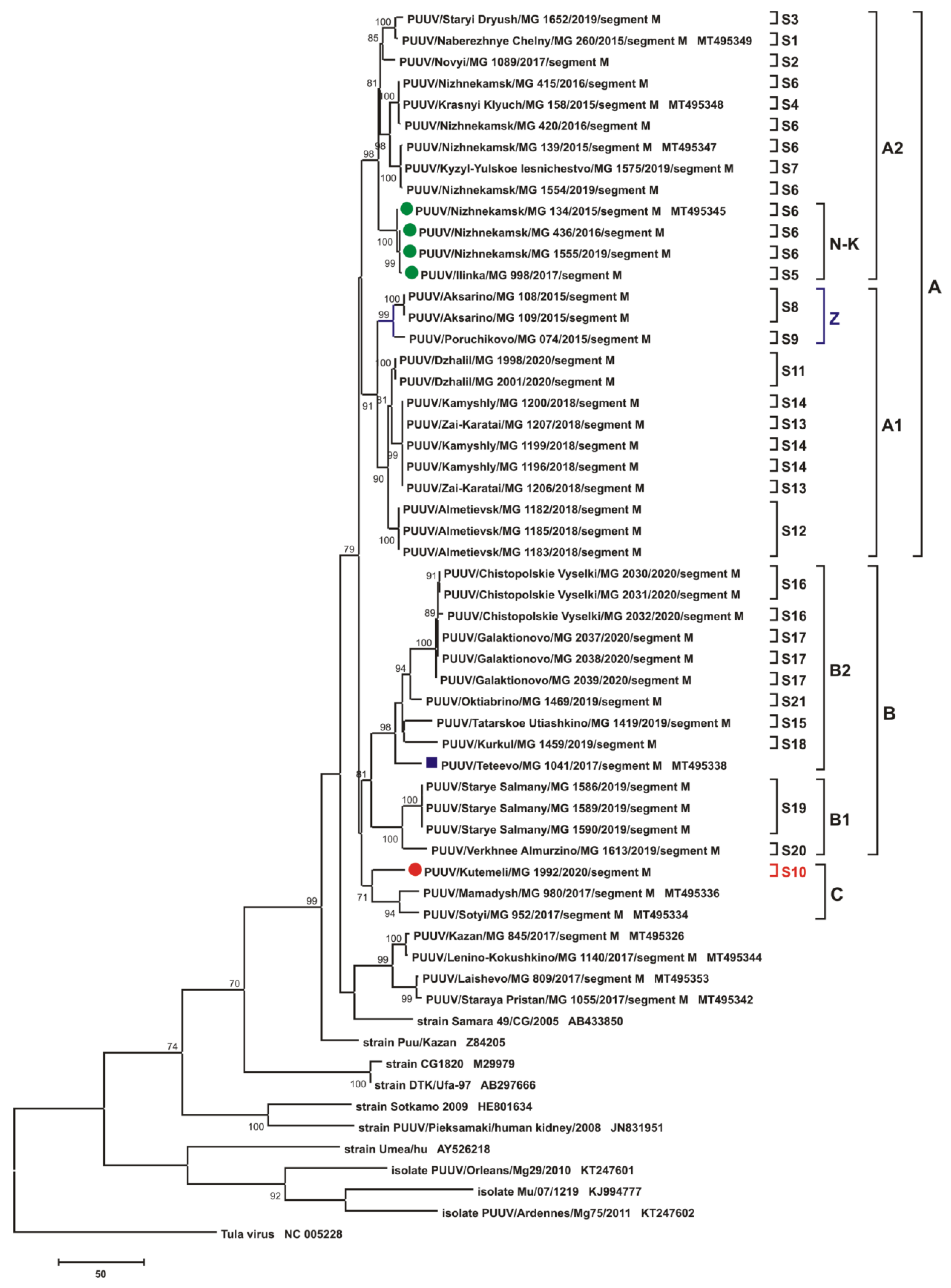
3.2.2. Comparison of the Partial M Segment Nucleotide and Glycoprotein Precursor Amino Acid Sequences of PUUV
3.2.3. Comparison of the Partial L Segment Nucleotide and RdRp Amino Acid Sequences of PUUV
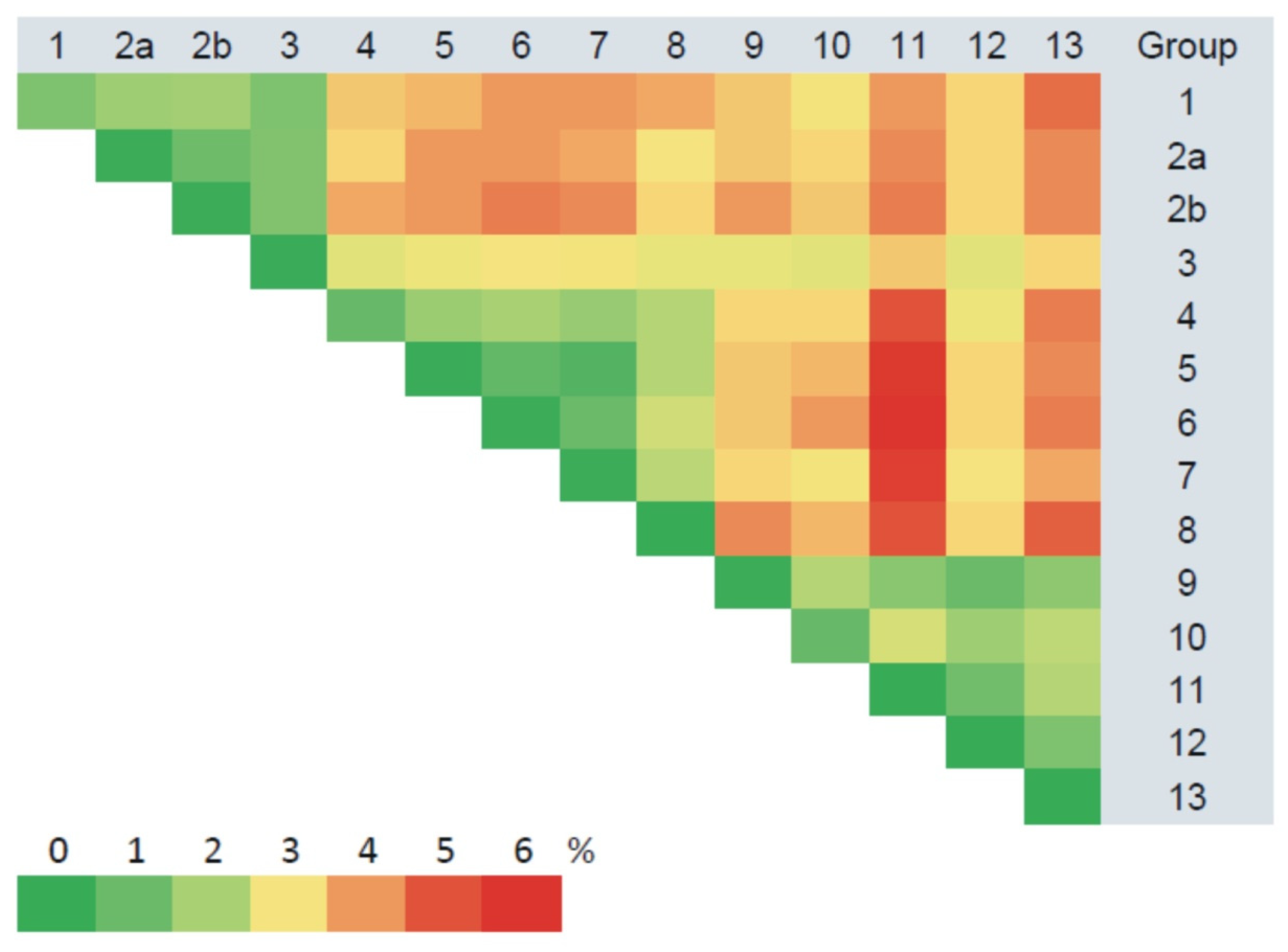
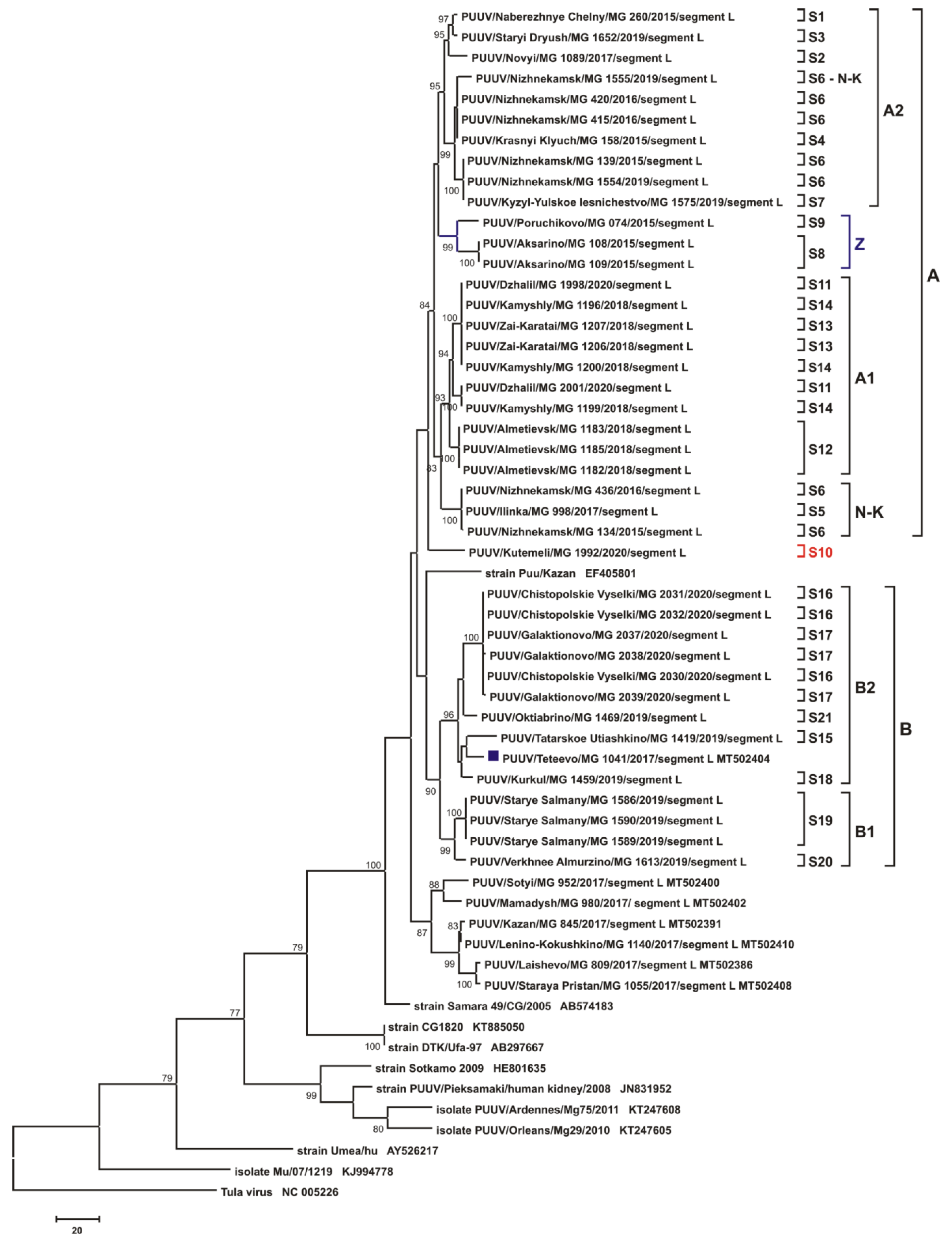
3.3. Phylogenetic Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Milholland, M.T.; Castro-Arellano, I.; Suzán, G.; Peña, G.E.G.; Lee, T.E.; Rohde, R.E.; Aguirre, A.A.; Mills, J.N. Global diversity and distribution of hantaviruses and their hosts. EcoHealth 2018, 15, 163–208. [Google Scholar] [CrossRef] [PubMed]
- Manigold, T.; Vial, P. Human hantavirus infections: Epidemiology, clinical features, pathogenesis and immunology. Swiss Med. Wkly. 2014, 144, w13937. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, J.; Hofmann, J.; Enders, M.; Tewald, F.; Oehme, R.M.; Rosenfeld, U.M.; Ali, H.S.; Schlegel, M.; Essbauer, S.; Osterberg, A.; et al. Multiple synchronous outbreaks of Puumala virus, Germany, 2010. Emerg. Infect. Dis. 2012, 18, 1461. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Calisher, C.; Schmaljohn, C. Manual of Hemorrhagic Fever with Renal Syndrome and Hantavirus Pulmonary Syndrome; WHO Collaborating Center for Virus Reference and Research (Hantaviruses); Asan Institute for Life Sciences: Seoul, Korea, 1998. [Google Scholar]
- Rospotrebnadzor. Prevention of Hemorrhagic Fever with Renal Syndrome (HFRS). 2021. Available online: https://www.rospotrebnadzor.ru/about/info/news/news_details.php?ELEMENT_ID=17714 (accessed on 24 July 2021).
- Korneev, M.G.; Chekashov, V.N.; Ivanova, A.V.; Matrosov, A.N.; Kuznetsov, A.A.; Shylov, M.M. Review of the Number of Carriers and Vectors of Zoonoses, Epizootic and Epidemiological Situation in the Volga Federal District in Second Half 2019 and Forecast for 2020. 2019. Available online: http://www.microbe.ru/files/PFO_revII_2019_prognI_2020.pdf (accessed on 24 July 2021).
- Vetter, P.; L’Huillier, A.G.; Montalbano, M.F.; Pigny, F.; Eckerle, I.; Torriani, G.; Rothenberger, S.; Laubscher, F.; Cordey, S.; Kaiser, L.; et al. Puumala virus infection in family, Switzerland. Emerg. Infect. Dis. 2021, 27, 658. [Google Scholar] [CrossRef] [PubMed]
- Garanina, E.; Martynova, E.; Davidyuk, Y.; Kabwe, E.; Ivanov, K.; Titova, A.; Markelova, M.; Zhuravleva, M.; Cherepnev, G.; Shakirova, V.G.; et al. Cytokine storm combined with humoral immune response defect in fatal hemorrhagic fever with renal syndrome case, Tatarstan, Russia. Viruses 2019, 11, 601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plyusnina, A.; Razzauti, M.; Sironen, T.; Niemimaa, J.; Vapalahti, O.; Vaheri, A.; Henttonen, H.; Plyusnin, A. Analysis of complete Puumala virus genome, Finland. Emerg. Infect. Dis. 2012, 18, 2070. [Google Scholar] [CrossRef] [PubMed]
- Vapalahti, O.; Mustonen, J.; Lundkvist, Å.; Henttonen, H.; Plyusnin, A.; Vaheri, A. Hantavirus infections in Europe. Lancet Infect. Dis. 2003, 3, 653–661. [Google Scholar] [CrossRef]
- Khismatullina, N.; Karimov, M.M.; Khaertynov, K.; Shuralev, E.; Morzunov, S.P.; Khaertynova, I.M.; Rizvanov, A.A.; Milova, I.V.; Khakimzyanova, M.B.; Sayfullina, G.S.; et al. Epidemiological dynamics of nephropathia epidemica in the Republic of Tatarstan, Russia, during the period of 1997–2013. Epidemiol. Infect. 2016, 144, 618–626. [Google Scholar] [CrossRef] [Green Version]
- De Vries, A.; Vennema, H.; Bekker, D.L.; Maas, M.; Adema, J.; Opsteegh, M.; Van Der Giessen, J.W.B.; Reusken, C.B.E.M. Characterization of Puumala hantavirus in bank voles from two regions in the Netherlands where human cases occurred. J. Gen. Virol. 2016, 97, 1500–1510. [Google Scholar] [CrossRef] [PubMed]
- Vapalahti, O.; Kallio-Kokko, H.; Salonen, E.-M.; Brummer-Korvenkontio, M.; Vaheri, A. Cloning and sequencing of Puumala virus Sotkamo strain S and M RNA segments: Evidence for strain variation in hantaviruses and expression of the nucleocapsid protein. J. Gen. Virol. 1992, 73, 829–838. [Google Scholar] [CrossRef]
- Jääskeläinen, K.M.; Kaukinen, P.; Minskaya, E.S.; Plyusnina, A.; Vapalahti, O.; Elliott, R.M.; Weber, F.; Vaheri, A.; Plyusnin, A. Tula and Puumala hantavirus NSs ORFs are functional and the products inhibit activation of the interferon-beta promoter. J. Med. Virol. 2007, 79, 1527–1536. [Google Scholar] [CrossRef] [PubMed]
- Giebel, L.B.; Stohwasser, R.; Zöller, L.; Bautz, E.K.; Darai, G. Determination of the coding capacity of the M genome segment of nephropathia epidemica virus strain Hällnäs B1 by molecular cloning and nucleotide sequence analysis. Virology 1989, 172, 498–505. [Google Scholar] [CrossRef]
- Kukkonen, S.K.; Vaheri, A.; Plyusnin, A. L protein, the RNA-dependent RNA polymerase of hantaviruses. Arch. Virol. 2005, 150, 533–556. [Google Scholar] [CrossRef]
- Castel, G.; Chevenet, F.; Razzauti, M.; Murri, S.; Marianneau, P.; Cosson, J.-F.; Tordo, N.; Plyusnin, A. Phylogeography of Puumala orthohantavirus in Europe. Viruses 2019, 11, 679. [Google Scholar] [CrossRef] [Green Version]
- Razzauti, M.; Plyusnina, A.; Niemimaa, J.; Henttonen, H.; Plyusnin, A. Co-circulation of two Puumala hantavirus lineages in Latvia: A Russian lineage described previously and a novel Latvian lineage. J. Med. Virol. 2012, 84, 314–318. [Google Scholar] [CrossRef]
- Davidyuk, Y.; Shamsutdinov, A.; Kabwe, E.; Ismagilova, R.; Martynova, E.; Belyaev, A.; Shuralev, E.; Trifonov, V.; Savitskaya, T.; Isaeva, G.; et al. Prevalence of the Puumala orthohantavirus strains in the pre-kama area of the Republic of Tatarstan, Russia. Pathogens 2020, 9, 540. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Ali, H.S.; Drewes, S.; de Melo, V.W.; Schlegel, M.; Freise, J.; Groschup, M.H.; Heckel, G.; Ulrich, R.G. Complete genome of a Puumala virus strain from Central Europe. Virus Genes 2015, 50, 292–298. [Google Scholar] [CrossRef]
- White, T.A.; Lundy, M.G.; Montgomery, W.I.; Montgomery, S.; Perkins, S.E.; Lawton, C.; Meehan, J.M.; Hayden, T.J.; Heckel, G.; Reid, N.; et al. Range expansion in an invasive small mammal: Influence of life-history and habitat quality. Biol. Invasions 2012, 14, 2203–2215. [Google Scholar] [CrossRef]
- Razzauti, M.; Plyusnina, A.; Sironen, T.; Henttonen, H.; Plyusnin, A. Analysis of Puumala hantavirus in a bank vole population in northern Finland: Evidence for co-circulation of two genetic lineages and frequent reassortment between strains. J. Gen. Virol. 2009, 90, 1923–1931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razzauti, M.; Plyusnina, A.; Henttonen, H.; Plyusnin, A. Accumulation of point mutations and reassortment of genomic RNA segments are involved in the microevolution of Puumala hantavirus in a bank vole (Myodes glareolus) population. J. Gen. Virol. 2008, 89, 1649–1660. [Google Scholar] [CrossRef]
- Dekonenko, A.; Yakimenko, V.; Ivanov, A.; Morozov, V.; Nikitin, P.; Khasanova, S.; Dzagurova, T.; Tkachenko, E.; Schmaljohn, C. Genetic similarity of Puumala viruses found in Finland and western Siberia and of the mitochondrial DNA of their rodent hosts suggests a common evolutionary origin. Infect. Genet. Evol. 2003, 3, 245–257. [Google Scholar] [CrossRef]
- Razzauti Sanfeliu, M. Microevolution of Puumala Hantavirus in Its Host, the Bank Vole (Myodes glareolus). Ph.D. Thesis, University of Helsinki, Helsinki, Finland, 2012. [Google Scholar]
- Khalil, H.; Ecke, F.; Evander, M.; Bucht, G.; Hörnfeldt, B. Population dynamics of bank voles predicts human puumala hantavirus risk. EcoHealth 2019, 16, 545–557. [Google Scholar] [CrossRef] [Green Version]
- Kiseleva, N.V. Long-term population dynamics of the bank vole in the Ilmen Nature Reserve. Russ. J. Ecol. 2020, 51, 188–194. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trapping Site | Location | No. of Trapped Bank Voles | No. of RT-PCR Positive Voles | No. Sequences Used for Analysis (Segment) | ||
|---|---|---|---|---|---|---|
| S (1302) a | M (1014) a | L (665) a | ||||
| S1 | Naberezhnye Chelny | 10 | 3 | 1 | 1 | 1 |
| S2 | Novyi | 17 | 5 | 1 | 1 | 1 |
| S3 | Staryi Dryush | 7 | 2 | 1 | 1 | 1 |
| S4 | Krasnyi Klyuch | 18 | 5 | 3 | 3 | 3 |
| S5 | Ilinka | 15 | 2 | 1 | 1 | 1 |
| S6 | Nizhnekamsk | 21 | 8 | 5 | 5 | 5 |
| S7 | Kyzyl-Yulskoe lesnichestvo | 10 | 1 | 1 | 1 | 1 |
| S8 | Aksarino | 3 | 3 | 2 | 2 | 2 |
| S9 | Poruchikovo | 5 | 5 | 1 | 1 | 1 |
| S10 | Kutemeli | 5 | 1 | 1 | 1 | 1 |
| S11 | Dzhalil | 5 | 2 | 2 | 2 | 2 |
| S12 | Almetievsk | 4 | 3 | 3 | 3 | 3 |
| S13 | Zai-Karatai | 4 | 3 | 2 | 2 | 2 |
| S14 | Kamyshly | 6 | 3 | 3 | 3 | 3 |
| S15 | Tatarskoe Utiashkino | 1 | 1 | 1 | 1 | 1 |
| S16 | Chistopolskie Vyselki | 5 | 3 | 3 | 3 | 3 |
| S17 | Galaktionovo | 5 | 3 | 3 | 3 | 3 |
| S18 | Kurkul | 3 | 1 | 1 | 1 | 1 |
| S19 | Starye Salmany | 8 | 3 | 3 | 3 | 3 |
| S20 | Verkhnee Almurzino | 8 | 1 | 1 | 1 | 1 |
| S21 | Oktiabrino | 2 | 1 | 1 | 1 | 1 |
| Total | 162 | 59 | 40 | 40 | 40 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davidyuk, Y.N.; Kabwe, E.; Shamsutdinov, A.F.; Knyazeva, A.V.; Martynova, E.V.; Ismagilova, R.K.; Trifonov, V.A.; Savitskaya, T.A.; Isaeva, G.S.; Urbanowicz, R.A.; et al. The Distribution of Puumala orthohantavirus Genome Variants Correlates with the Regional Landscapes in the Trans-Kama Area of the Republic of Tatarstan. Pathogens 2021, 10, 1169. https://doi.org/10.3390/pathogens10091169
Davidyuk YN, Kabwe E, Shamsutdinov AF, Knyazeva AV, Martynova EV, Ismagilova RK, Trifonov VA, Savitskaya TA, Isaeva GS, Urbanowicz RA, et al. The Distribution of Puumala orthohantavirus Genome Variants Correlates with the Regional Landscapes in the Trans-Kama Area of the Republic of Tatarstan. Pathogens. 2021; 10(9):1169. https://doi.org/10.3390/pathogens10091169
Chicago/Turabian StyleDavidyuk, Yuriy N., Emmanuel Kabwe, Anton F. Shamsutdinov, Anna V. Knyazeva, Ekaterina V. Martynova, Ruzilya K. Ismagilova, Vladimir A. Trifonov, Tatiana A. Savitskaya, Guzel S. Isaeva, Richard A. Urbanowicz, and et al. 2021. "The Distribution of Puumala orthohantavirus Genome Variants Correlates with the Regional Landscapes in the Trans-Kama Area of the Republic of Tatarstan" Pathogens 10, no. 9: 1169. https://doi.org/10.3390/pathogens10091169
APA StyleDavidyuk, Y. N., Kabwe, E., Shamsutdinov, A. F., Knyazeva, A. V., Martynova, E. V., Ismagilova, R. K., Trifonov, V. A., Savitskaya, T. A., Isaeva, G. S., Urbanowicz, R. A., Khaiboullina, S. F., Rizvanov, A. A., & Morzunov, S. P. (2021). The Distribution of Puumala orthohantavirus Genome Variants Correlates with the Regional Landscapes in the Trans-Kama Area of the Republic of Tatarstan. Pathogens, 10(9), 1169. https://doi.org/10.3390/pathogens10091169