
Plasmodium vivax Genetic Diversity in Panama: Challenges for Malaria Elimination in Mesoamerica

 , and
, and
Abstract
1. Introduction
2. Results
2.1. Genotyping of Csp, Msp-1, and Msp-3α Genes by PCR-RFLP
2.2. Multiple Correspondence Analysis
2.3. Csp Sequencing and Phylogenetic Analysis
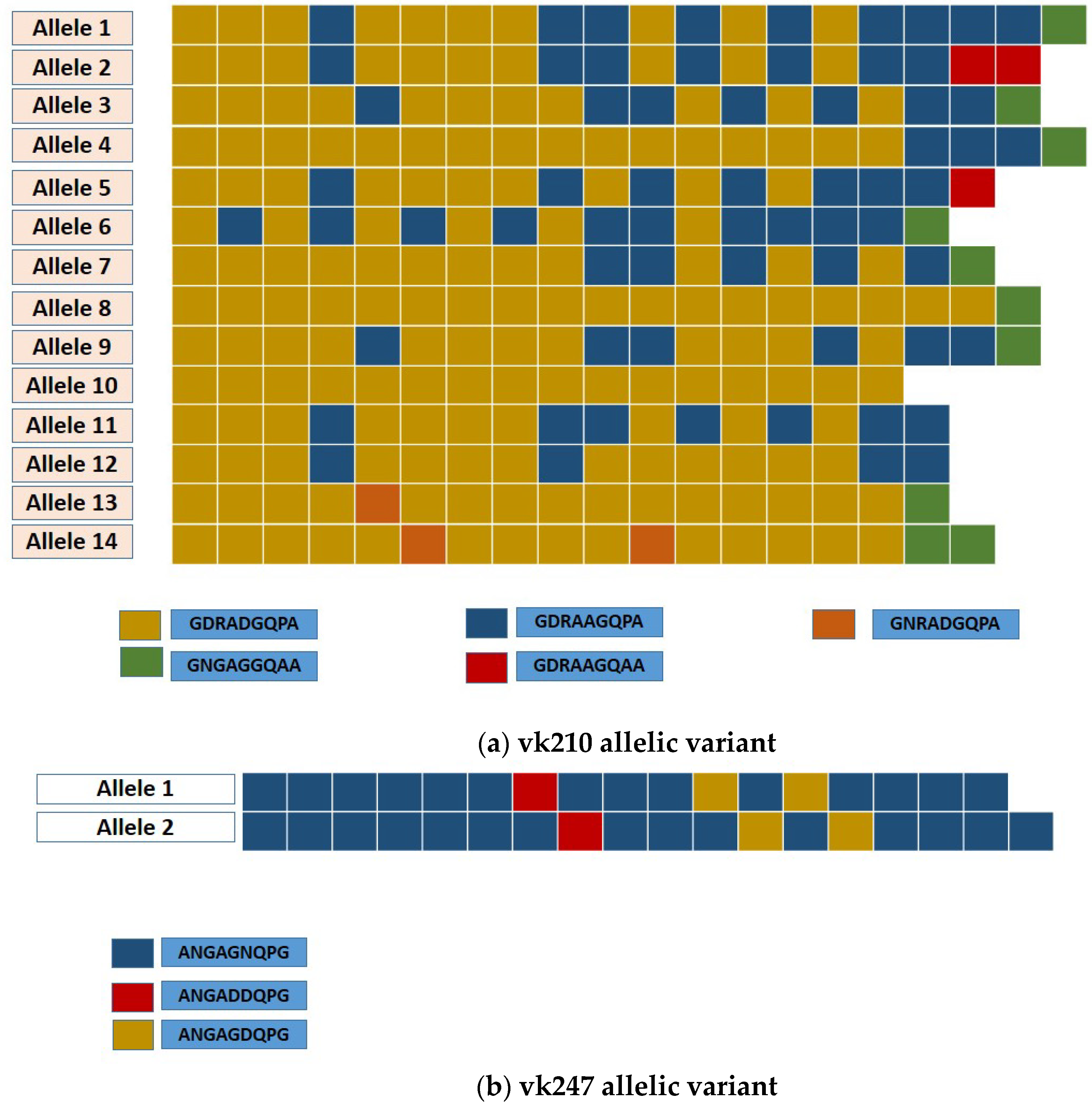
2.4. Analysis of Csp Allelic Variants
3. Discussion
4. Materials and Methods
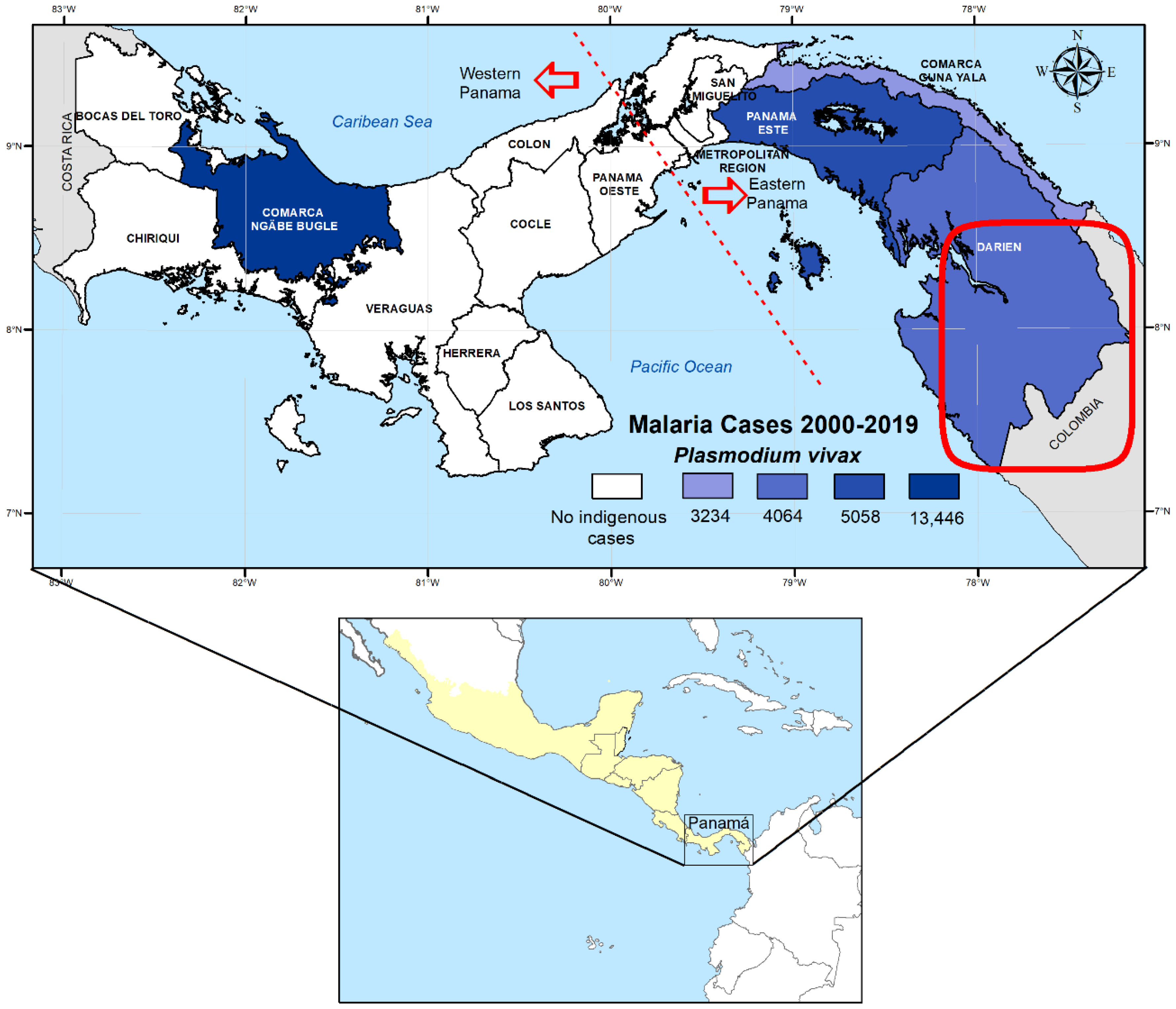
4.1. Study Site
4.2. Sample Collection and Plasmodium Diagnosis
4.3. Plasmodium Vivax Genotyping and Csp Phylogenetic Analysis
4.4. Data and Statistical Analysis
4.5. Ethical Statement
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pan American Health Organization. Mesoamerican Master Plans. Technical Cooperation Agreement between the Ministry of Foreign Affairs of Mexico and the Pan American Health Organization=Planes Maestros Mesoamericanos. Acuerdo de Cooperación Técnica entre la Secretaría de Relaciones Exteriores de México y la Organización Panamericana de la Salud. 2016. Available online: http://www.proyectomesoamerica.org:8088/smsp/phocadownload/Estrategico/MESOAMERICAN_MASTER_PLAN%20for%20web.pdf (accessed on 10 January 2021).
- Consejo de Ministros de Salud de Centroamérica y República Dominicana; Declaración-Hacia la Eliminación de La Malaria en Mesoamérica y La Isla de La Española En El 2020. COMISCA XROd: San José, Costa Rica, 2013. Available online: https://www.google.com.hk/url?sa=t&rct=j&q=&esrc=s&source=web&cd=&ved=2ahUKEwiIi4Wdw5nyAhUXA4gKHV8bDp0QFnoECAQQAQ&url=https%3A%2F%2Fwww.sica.int%2Fdownload%2F%3F79241&usg=AOvVaw0GZR-ORn8gYrJUDMPzN9pd (accessed on 5 January 2021).
- World Health Organization. World Malaria Report 2020: 20 Years of Global Progress and Challenges. 2020; (WHO/CDS/GMP/2018.13). Licence: CC BY-NC-SA 3.0 IGO. Available online: https://www.who.int/teams/global-malariaprogramme/reports/world-malaria-report-2020 (accessed on 10 January 2021).
- El Salvador Certified as Malaria-Free by WHO. Available online: https://www.who.int/news/item/25-02-2021-el-salvador-certified-as-malaria-free-by-who (accessed on 10 January 2021).
- World Health Organization. Update on the E-2020 Initiative of 21 Malaria-Eliminating Countries: Report and Country Briefs; World Health Organization: Geneva, Switzerland, 2018. Available online: https://apps.who.int/iris/bitstream/handle/10665/273633/WHO-CDS-GMP-2018.13-eng.pdf?sequence=1&isAllowed=y (accessed on 10 January 2021).
- Hotez, P.J.; Woc-Colburn, L.; Bottazzi, M.E. Neglected tropical diseases in Central America and Panama: Review of their prevalence, populations at risk and impact on regional development. Int. J. Parasitol. 2014, 44, 597–603. [Google Scholar] [CrossRef]
- Chaves, L.F.; Ramírez Rojas, M.; Prado, M.; Garcés, J.L.; Salas Peraza, D.; Marín Rodríguez, R. Health policy impacts on malaria transmission in Costa Rica. Parasitology 2020, 147, 999–1007. [Google Scholar] [CrossRef]
- Hotez, P.J.; Damania, A.; Bottazzi, M.E. Central Latin America: Two decades of challenges in neglected tropical disease control. PLoS Negl. Trop. Dis. 2020, 14, e0007962. [Google Scholar] [CrossRef]
- Ministerio de Salud. Plan Estratégico de Eliminación de la Malaria (PEEM) en Panamá 2018–2022; Ministerio de Salud de Panamá: Panamá, Panama, 2018; pp. 8–39.
- Hurtado, L.; Cumbrera, A.; Rigg, C.; Perea, M.; Santamaría, A.M.; Chaves, L.F.; Moreno, D.; Romero, L.; Lasso, J.; Caceres, L.; et al. Long-term transmission patterns and public health policies leading to malaria elimination in Panamá. Malar. J. 2020, 19, 265. [Google Scholar] [CrossRef] [PubMed]
- Hurtado, L.A.; Cáceres, L.; Chaves, L.F.; Calzada, J.E. When climate change couples social neglect: Malaria dynamics in Panamá. Emerg. Microbes Infect. 2014, 3, 4. [Google Scholar] [CrossRef]
- Obaldia, N., 3rd. Determinants of low socio-economic status and risk of Plasmodium vivax malaria infection in Panama (2009-2012): A case-control study. Malar. J. 2015, 14, 14. [Google Scholar] [CrossRef] [PubMed]
- The World Bank in Panama. 2020. Available online: https://www.worldbank.org/en/country/panama/overview (accessed on 21 January 2020).
- Servicio Nacional de Migración de Panamá. Movimiento Migratorio. 2019. Available online: https://www.migracion.gob.pa/images/pdf/IRREGULARES_POR%2520_DARIEN_DICIEMBRE_2019.pdf (accessed on 2 July 2019).
- Santamaría, A.M.; Vásquez, V.; Rigg, C.; Moreno, D.; Romero, L.; Justo, C.; Chaves, L.F.; Saldaña, A.; Calzada, J.E. Plasmodium falciparum Genetic Diversity in Panamá Based on glurp, msp-1 and msp-2 Genes: Implications for Malaria Elimination in Mesoamerica. Life 2020, 28, 319. [Google Scholar] [CrossRef] [PubMed]
- Brito, C.F.; Ferreira, M.U. Molecular markers and genetic diversity of Plasmodium vivax. Memórias do Inst. Oswaldo Cruz 2011, 106, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Instituto Geográfico Nacional Tommy Guardia. Atlas Nacional de la República de Panamá 2015; Instituto Geográfico Nacional Tommy Guardia: Panamá, Panama, 2016; ISBN 978-9962-11-048-4. [Google Scholar]
- Arnott, A.; Barnadas, C.; Senn, N.; Siba, P.; Mueller, I.; Reeder, J.C.; Barry, A.E. High genetic diversity of Plasmodium vivax on the north coast of Papua New Guinea. Am. J. Trop. Med. Hyg. 2013, 89, 188–194. [Google Scholar] [CrossRef]
- Medicines for Malaria Venture and World Health Organization. Methods and Techniques for Clinical Trials on Antimalarial Drug Efficacy: Genotyping to Identify Parasite Populations; Informal Consultation Organized by the Medicines for Malaria Venture and Cosponsored; World Health Organization: Amsterdam, The Netherlands, 2007; p. 45. [Google Scholar]
- De Souza, A.M.; de Araújo, F.C.F.; Fontes, C.J.F.; Carvalho, L.H.; de Brito, C.F.A.; de Sousa, T.N. Multiple-clone infections of Plasmodium vivax: Definition of a panel of markers for molecular epidemiology. Malar. J. 2015, 14, 330. [Google Scholar] [CrossRef] [PubMed]
- Chaves, L.F.; Huber, J.H.; Rojas Salas, O.; Ramírez Rojas, M.; Romero, L.M.; Gutiérrez Alvarado, J.M.; Perkins, T.A.; Prado, M.; Marín Rodríguez, R. Malaria Elimination in Costa Rica: Changes in Treatment and Mass Drug Administration. Microorganisms 2020, 8, 984. [Google Scholar] [CrossRef]
- Yates, C. As More Migrants from Africa and Asia Arrive in Latin America, Governments Seek Orderly and Controlled Pathways. Available online: https://www.migrationpolicy.org/article/extracontinental-migrants-latin-america (accessed on 22 October 2019).
- Ocampo González, M.; Arboleda Cardona, S. Colombia y los flujos mixtos de migrantes en el derecho internacional de los refugiados. Rev. Opinión Jurídica 2016, 15, 93–108. Available online: https://doi.org/10.22395/ojum.v15n30a4 (accessed on 22 October 2019). [CrossRef]
- González-Cerón, L.; Martinez-Barnetche, J.; Montero-Solís, C.; Santillán, F.; Soto, A.M.; Rodríguez, M.H.; Espinosa, B.J.; Chávez, O.A. Molecular epidemiology of Plasmodium vivax in Latin America: Polymorphism and evolutionary relationships of the circumsporozoite gene. Malar. J. 2013, 12, 243. [Google Scholar] [CrossRef]
- González-Cerón, L.; Rodríguez, M.H.; Nettel-Cruz, J.A.; Hernández-Ávila, J.E.; Malo-García, I.R.; Santillán-Valenzuela, F.; Villarreal-Treviño, C. Plasmodium vivax CSP-Pvs25 variants from southern Mexico produce distinct patterns of infectivity for Anopheles albimanus versus An. pseudopunctipennis, in each case independent of geographical origin. Parasit Vectors 2019, 20, 86. [Google Scholar] [CrossRef]
- Lopez, A.C.; Ortiz, A.; Coello, J.; Sosa-Ochoa, W.; Torres, R.E.; Banegas, E.I.; Jovel, I.; Fontecha, G.A. Genetic diversity of Plasmodium vivax and Plasmodium falciparum in Honduras. Malar. J. 2012, 26, 391. [Google Scholar] [CrossRef]
- Mendizábal-Cabrera, R.; Padilla, N. Diversidad genética de Plasmodium vivax en regiones de alto riesgo de malaria en Guatemala. Rev. de la Univ. del Val. Guatem. 2006, 15, 62–79. [Google Scholar]
- Gonzalez, J.M.; Hurtado, S.; Arévalo-Herrera, M.; Herrera, S. Variants of the Plasmodium vivax circumsporozoite protein (VK210 and VK247) in Colombian isolates. Mem. Inst. Oswaldo Cruz 2001, 96, 709–712. [Google Scholar] [CrossRef]
- Hernández-Martínez, M.Á.; Escalante, A.A.; Arévalo-Herrera, M.; Herrera, S. Antigenic diversity of the Plasmodium vivax circumsporozoite protein in parasite isolates of Western Colombia. Am. J. Trop. Med. Hyg. 2011, 84, 51–57. [Google Scholar] [CrossRef]
- Arnott, A.; Barry, A.E.; Reeder, J.C. Understanding the population genetics of Plasmodium vivax is essential for malaria control and elimination. Malar. J. 2012, 11, 14. [Google Scholar] [CrossRef]
- Loaiza, J.R.; Bermingham, E.; Scott, M.E.; Rovira, J.R.; Conn, J.E. Species composition and distribution of adult Anopheles (Diptera: Culicidae) in Panama. J. Med. Entomol. 2008, 45, 841–851. [Google Scholar] [CrossRef]
- Torres-Cosme, R.; Rigg, C.; Santamaria, A.M.; Vasquez, V.; Victoria, C.; Ramirez, J.L.; Calzada, J.E.; Caceres, L. Natural malaria infection in anophelines vectors and their incrimination in local malaria transmission in Darien, Panama. PLoS ONE 2021, 16, e0250059. [Google Scholar] [CrossRef] [PubMed]
- Grieco, J.P.; Johnson, S.; Achee, L.N.; Masuoka, P.; Pope, K.; Rejmankova, E.; Vanzie, F.; Andre, R.; Roberts, D. Distribution of Anopheles albimanus, Anopheles vestitipennis and Anopheles crucians associated with land use in northern Belize. J. Med. Entomol. 2006, 43, 614–622. [Google Scholar] [CrossRef]
- Loyola, E.G.; Arredondo, J.I.; Rodriguez, M.H.; Brown, D.N.; Vaca-Marin, M.A. Anopheles vestitipennis, the probable vector of Plasmodium vivax in the Lacandon forest of Chiapas, Mexico. Trans. R. Soc. Trop. Med. Hyg. 1991, 85, 171–174. [Google Scholar] [CrossRef]
- Escobar, D.; Ascencio, K.; Ortiz, A.; Palma, A.; Fontecha, G. Distribution and phylogenetic diversity of Anopheles species in malaria endemic areas of Honduras in an elimination setting. Parasit Vectors 2020, 13, 333. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Ceron, L.; Rodriguez, M.H.; Nettel, J.C.; Villarreal, C.; Kain, K.C.; Hernandez, J.E. Differential susceptibilities of Anopheles albimanus and Anopheles pseudopunctipennis to infections with coindigenous Plasmodium vivax variants VK210 and VK247 in southern Mexico. Infect. Immun. 1999, 67, 410–412. [Google Scholar] [CrossRef]
- Cáceres Carrera, L.; Victoria, C.; Ramirez, J.L.; Jackman, C.; Calzada, J.E.; Torres, R. Study of the epidemiological behavior of malaria in the Darien Region, Panama, 2015–2017. PLoS ONE 2019, 15, e0224508. [Google Scholar] [CrossRef] [PubMed]
- Buyon, L.E.; Santamaria, A.M.; Early, A.M.; Quijada, M.; Barahona, I.; Lasso, J.; Avila, M.; Volkman, S.K.; Marti, M.; Neafsey, D.E.; et al. Population genomics of Plasmodium vivax in Panama to assess the risk of case importation on malaria elimination. PLoS Negl. Trop. Dis. 2020, 14, e0008962. [Google Scholar] [CrossRef]
- Rodriguez-Morales, A.J.; Delgado, L.; Martinez, N.; Franco-Paredes, C. Impact of imported malaria on the burden of disease in northeastern Venezuela. J. Travel Med. 2006, 13, 15–20. [Google Scholar] [CrossRef]
- Cui, L.; Yan, G.; Sattabongkot, J.; Cao, Y.; Chen, B.; Chen, X.; Fan, Q.; Jongwutiwes, S.; Parker, D.; Sirichaisinthop, J.; et al. Malaria in the Greater Mekong Subregion: Heterogeneity and complexity. Acta Trop. 2012, 121, 227–239. [Google Scholar] [CrossRef]
- Ministerio de Salud Panamá. Manual de Normas y Procedimientos para Malaria; Ministerio de Salud Panamá: Panamá, Panama, 2011; pp. 36–317.
- Kotepui, M.; Piwkham, D.; PhunPhuech, B.; Phiwklam, N.; Chupeerach, C.; Duangmano, S. Effects of malaria parasite density on blood cell parameters. PLoS ONE 2015, 10, 3. [Google Scholar] [CrossRef]
- Snounou, G.; Viriyakosol, S.; Jarra, W.; Thaithong, S.; Brown, K.N. Identification of the four human malaria parasite species in field samples by the polymerase chain reaction and detection of a high prevalence of mixed infections. Mol. Biochem. Parasitol. 1993, 58, 283–292. [Google Scholar] [CrossRef]
- Imwong, M.; Pukrittayakamee, S.; Grüner, A.C.; Rénia, L.; Letourneur, F.; Looareesuwan, S.; White, N.J.; Snounou, G. Practical PCR genotyping protocols for Plasmodium vivax using Pvcs and Pvmsp1. Malar. J. 2005, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Bruce, M.C.; Galinski, M.R.; Barnwell, J.W.; Snounou, G.; Day, K.P. Polymorphism at the merozoite surface protein-3alpha locus of Plasmodium vivax: Global and local diversity. Am. J. Trop. Med. Hyg. 1999, 61, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Rigg, C.A.; Calzada, J.E.; Saldaña, A.; Perea, M.; Chaves, L.F.; Valderrama, A. Leishmania spp. Infection Rate and Feeding Patterns of Sand Flies (Diptera: Psychodidae) from a Hyperendemic Cutaneous Leishmaniasis Community in Panamá. Am. J. Trop. Med. Hyg. 2019, 100, 798–807. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Csp Vk210 | Csp Vk247 | Msp-1 Genotype G1 | Msp-1 Genotype G2 | Msp-1 Genotype G3 | Msp-1 Genotype G4 | Msp-3α Genotype G1 | Msp-3α Genotype G2 | Msp-3α Genotype G3 | |
|---|---|---|---|---|---|---|---|---|---|
| Bocas del Toro | 12 (5.8%) | 0 | 15 (6.4%) | 4 (1.7%) | 0 | 0 | 13 (5.5%) | 0 | 0 |
| Veraguas | 25 (12%) | 0 | 25 (10.7%) | 0 | 0 | 0 | 25 (10.6%) | 0 | 0 |
| Panamá Metro | 17 (8.2%) | 0 | 15 (6.4%) | 1 (0.4%) | 0 | 0 | 16 (6.8%) | 0 | 0 |
| Panamá Este | 40 (19.2%) | 0 | 42 (17.9%) | 5 (2.1%) | 0 | 0 | 43 (18.2%) | 2 (1%) | 0 |
| Guna Yala | 29 (13.9%) | 5 (2.4%) | 22 (9.4%) | 3 (1.3%) | 0 | 0 | 15 (6.3%) | 11 (4.7%) | 0 |
| Darién | 35 (16.9%) | 29 (13.9%) | 48 (20.5%) | 23 (9.8%) | 0 | 0 | 53 (22.4%) | 25 (10.6%) | 1 (0.4%) |
| Imported | 12 (5.8%) | 4 (1.9%) | 10 (4.3%) | 18 (7.7%) | 1 (0.4%) | 2 (1%) | 26 (11%) | 5 (2.1%) | 1 (0.4%) |
| Total | 170 | 38 | 177 | 54 | 1 | 2 | 191 | 43 | 2 |
| Csp Genotype | Msp-1 Genotype | Msp-3α Genotype | Bocas del Toro | Veraguas | Panamá Metro | Panamá Este | Guna Yala | Darién | Imported | Total (%) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Haplotype 1 | vk210 | G2 | G1 | 1 | 4 | 5 (3.1) | |||||
| Haplotype 2 | vk247 | G2 | G2 | 1 | 1 | 2 (1.2) | |||||
| Haplotype 3 | vk247 | G2 | G1 | 1 | 1 (0.6) | ||||||
| Haplotype 4 | vk247 | G1 | G2 | 6 | 1 | 7 (4.3) | |||||
| Haplotype 5 | vk247 | G1 | G1 | 16 | 16 (9.8) | ||||||
| Haplotype 6 | vK210 | G1 | G2 | 7 | 7 (4.3) | ||||||
| Haplotype 7 | vK210 | G2 | G1 | 1 | 9 | 6 | 16 (9.8) | ||||
| Haplotype 8 | vk247 | G3 | G3 | 1 | 1 (0.6) | ||||||
| Haplotype 9 | vk247 | G4 | G1 | 1 | 1 (0.6) | ||||||
| Haplotype 10 | vk210 | G1 | G1 | 12 | 25 | 14 | 38 | 8 | 8 | 2 | 107 (65.6) |
| Total | 12 | 25 | 15 | 38 | 10 | 52 | 11 | 163 |
| Nonapeptide Variant | Isolate Origin | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Vk210 Subtype | A | B | C | D | E | WP | EP | Imp | Total | % |
| Allele 1 | 10 | 9 | 1 | 3 | 2 | 5 | 7.8 | |||
| Allele 2 | 10 | 7 | 2 | 1 | 1 | 1.6 | ||||
| Allele 3 | 11 | 7 | 1 | 7 | 27 | 8 | 42 | 65.6 | ||
| Allele 4 | 16 | 3 | 1 | 1 | 1 | 2 | 3.1 | |||
| Allele 5 | 10 | 7 | 1 | 1 | 1 | 1.6 | ||||
| Allele 6 | 6 | 10 | 1 | 1 | 1 | 1.7 | ||||
| Allele 7 | 12 | 5 | 1 | 1 | 1 | 1.6 | ||||
| Allele 8 | 18 | 1 | 1 | 1 | 1.6 | |||||
| Allele 9 | 12 | 6 | 1 | 1 | 1 | 2 | 3.1 | |||
| Allele 10 | 16 | 2 | 2 | 3.1 | ||||||
| Allele 11 | 10 | 7 | 2 | 2 | 3.1 | |||||
| Allele 12 | 13 | 4 | 2 | 2 | 3.1 | |||||
| Allele 13 | 15 | 1 | 1 | 1 | 1 | 1.6 | ||||
| Allele 14 | 14 | 2 | 2 | 1 | 1 | 1.6 | ||||
| Total | 12 | 34 | 18 | 64 | 100 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santamaría, A.M.; Vásquez, V.; Rigg, C.; Samudio, F.; Moreno, D.; Romero, L.; Saldaña, A.; Chaves, L.F.; Calzada, J.E. Plasmodium vivax Genetic Diversity in Panama: Challenges for Malaria Elimination in Mesoamerica. Pathogens 2021, 10, 989. https://doi.org/10.3390/pathogens10080989
Santamaría AM, Vásquez V, Rigg C, Samudio F, Moreno D, Romero L, Saldaña A, Chaves LF, Calzada JE. Plasmodium vivax Genetic Diversity in Panama: Challenges for Malaria Elimination in Mesoamerica. Pathogens. 2021; 10(8):989. https://doi.org/10.3390/pathogens10080989
Chicago/Turabian StyleSantamaría, Ana María, Vanessa Vásquez, Chystrie Rigg, Franklyn Samudio, Dianik Moreno, Luis Romero, Azael Saldaña, Luis Fernando Chaves, and José Eduardo Calzada. 2021. "Plasmodium vivax Genetic Diversity in Panama: Challenges for Malaria Elimination in Mesoamerica" Pathogens 10, no. 8: 989. https://doi.org/10.3390/pathogens10080989
APA StyleSantamaría, A. M., Vásquez, V., Rigg, C., Samudio, F., Moreno, D., Romero, L., Saldaña, A., Chaves, L. F., & Calzada, J. E. (2021). Plasmodium vivax Genetic Diversity in Panama: Challenges for Malaria Elimination in Mesoamerica. Pathogens, 10(8), 989. https://doi.org/10.3390/pathogens10080989