
TNF Decoy Receptors Encoded by Poxviruses

 , , and
, , and
Abstract
1. TNF Biology
2. Virally Encoded TNFRs and TNFBPs
2.1. Virally Encoded TNFRs
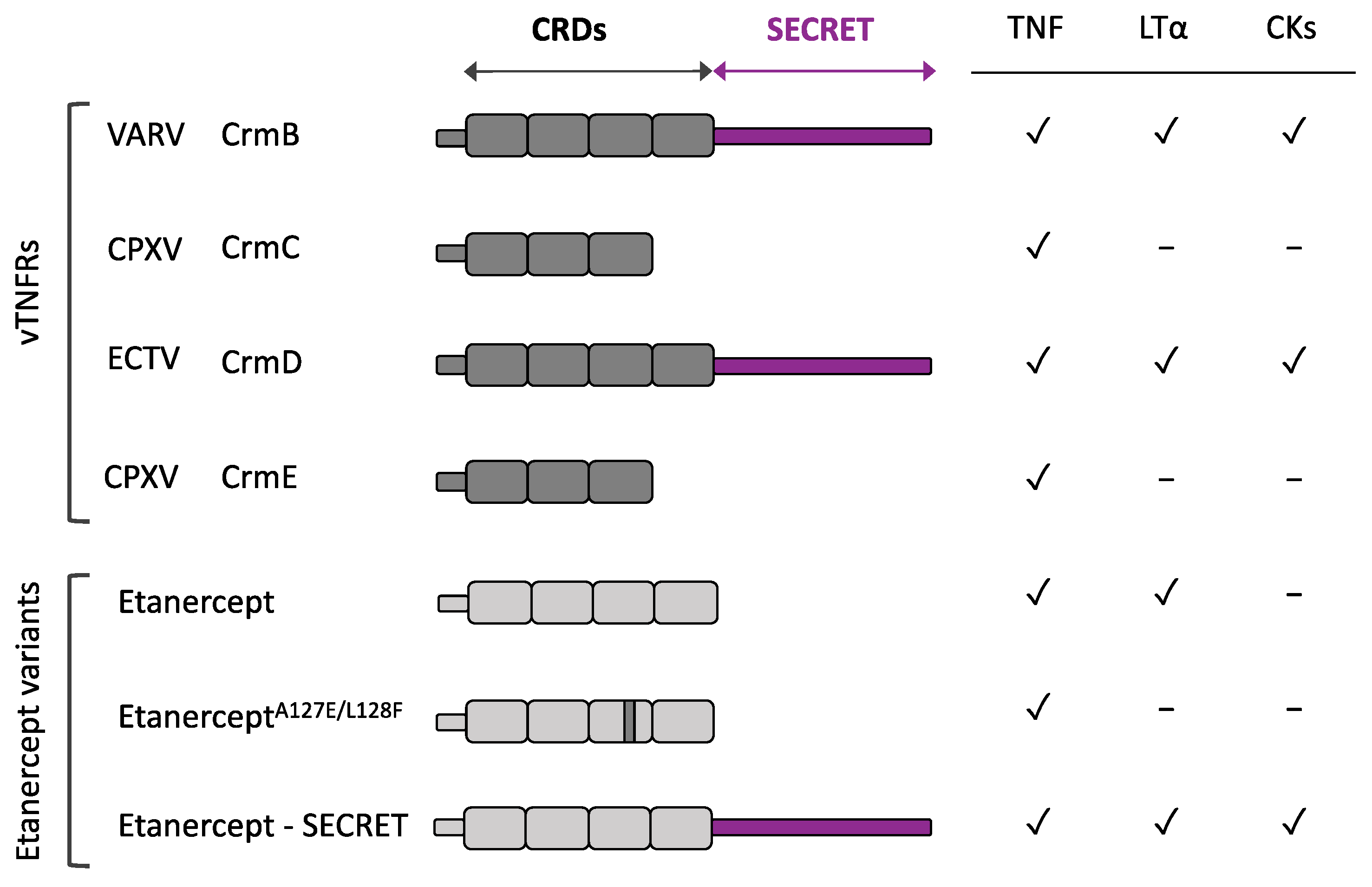
2.1.1. vTNFRs Affinities and Evolution
2.1.2. Modulation of tmTNF
2.1.3. Addition of a Chemokine Binding Domain: SECRET
2.1.4. T2 Protein
2.1.5. vCD30
2.2. vTNFBPs
3. Relevance in Poxvirus Pathogenesis
4. Therapeutic Use of vTNFRs
Author Contributions
Funding
Conflicts of Interest
References
- Watts, A.D.; Hunt, N.H.; Wanigasekara, Y.; Bloomfield, G.; Wallach, D.; Roufogalis, B.D.; Chaudhri, G. A Casein Kinase I Motif Present in the Cytoplasmic Domain of Members of the Tumour Necrosis Factor Ligand Family Is Implicated in “Reverse Signalling”. EMBO J. 1999, 18, 2119–2126. [Google Scholar] [CrossRef] [PubMed]
- Zelová, H.; Hošek, J. TNF-α Signalling and Inflammation: Interactions between Old Acquaintances. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. Al 2013, 62, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.; Calvano, S.E.; Lowry, S.F. Inflammatory Cytokines and Cell Response in Surgery. Surgery 2000, 127, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF Receptor Superfamilies: Integrating Mammalian Biology. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef]
- Aggarwal, B.B. Signalling Pathways of the TNF Superfamily: A Double-Edged Sword. Nat. Rev. Immunol. 2003, 3, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A Metalloproteinase Disintegrin That Releases Tumour-Necrosis Factor-Alpha from Cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef]
- Perez, C.; Albert, I.; DeFay, K.; Zachariades, N.; Gooding, L.; Kriegler, M. A Nonsecretable Cell Surface Mutant of Tumor Necrosis Factor (TNF) Kills by Cell-to-Cell Contact. Cell 1990, 63, 251–258. [Google Scholar] [CrossRef]
- Grell, M.; Douni, E.; Wajant, H.; Löhden, M.; Clauss, M.; Maxeiner, B.; Georgopoulos, S.; Lesslauer, W.; Kollias, G.; Pfizenmaier, K.; et al. The Transmembrane Form of Tumor Necrosis Factor Is the Prime Activating Ligand of the 80 KDa Tumor Necrosis Factor Receptor. Cell 1995, 83, 793–802. [Google Scholar] [CrossRef]
- Declercq, W.; Vandenabeele, P.; Fiers, W. Dimerization of Chimeric Erythropoietin/75 KDa Tumour Necrosis Factor (TNF) Receptors Transduces TNF Signals: Necessity for the 75 KDa-TNF Receptor Transmembrane Domain. Cytokine 1995, 7, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Levine, S.J. Molecular Mechanisms of Soluble Cytokine Receptor Generation. J. Biol. Chem. 2008, 283, 14177–14181. [Google Scholar] [CrossRef] [PubMed]
- Lainez, B.; Fernandez-Real, J.M.; Romero, X.; Esplugues, E.; Cañete, J.D.; Ricart, W.; Engel, P. Identification and Characterization of a Novel Spliced Variant That Encodes Human Soluble Tumor Necrosis Factor Receptor 2. Int. Immunol. 2004, 16, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; O’Rourke, K.; Chinnaiyan, A.M.; Gentz, R.; Ebner, R.; Ni, J.; Dixit, V.M. The Receptor for the Cytotoxic Ligand TRAIL. Science 1997, 276, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Pitti, R.M.; Marsters, S.A.; Lawrence, D.A.; Roy, M.; Kischkel, F.C.; Dowd, P.; Huang, A.; Donahue, C.J.; Sherwood, S.W.; Baldwin, D.T.; et al. Genomic Amplification of a Decoy Receptor for Fas Ligand in Lung and Colon Cancer. Nature 1998, 396, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Degli-Esposti, M.A.; Smolak, P.J.; Walczak, H.; Waugh, J.; Huang, C.P.; DuBose, R.F.; Goodwin, R.G.; Smith, C.A. Cloning and Characterization of TRAIL-R3, a Novel Member of the Emerging TRAIL Receptor Family. J. Exp. Med. 1997, 186, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Domonkos, A.; Udvardy, A.; László, L.; Nagy, T.; Duda, E. Receptor-like Properties of the 26 KDa Transmembrane Form of TNF. Eur. Cytokine Netw. 2001, 12, 411–419. [Google Scholar] [PubMed]
- Eissner, G.; Kolch, W.; Scheurich, P. Ligands Working as Receptors: Reverse Signaling by Members of the TNF Superfamily Enhance the Plasticity of the Immune System. Cytokine Growth Factor Rev. 2004, 15, 353–366. [Google Scholar] [CrossRef]
- Browning, J.L.; Ngam-ek, A.; Lawton, P.; DeMarinis, J.; Tizard, R.; Chow, E.P.; Hession, C.; O’Brine-Greco, B.; Foley, S.F.; Ware, C.F. Lymphotoxin Beta, a Novel Member of the TNF Family That Forms a Heteromeric Complex with Lymphotoxin on the Cell Surface. Cell 1993, 72, 847–856. [Google Scholar] [CrossRef]
- Medvedev, A.E.; Espevik, T.; Ranges, G.; Sundan, A. Distinct Roles of the Two Tumor Necrosis Factor (TNF) Receptors in Modulating TNF and Lymphotoxin Alpha Effects. J. Biol. Chem. 1996, 271, 9778–9784. [Google Scholar] [CrossRef] [PubMed]
- Mauri, D.N.; Ebner, R.; Montgomery, R.I.; Kochel, K.D.; Cheung, T.C.; Yu, G.L.; Ruben, S.; Murphy, M.; Eisenberg, R.J.; Cohen, G.H.; et al. LIGHT, a New Member of the TNF Superfamily, and Lymphotoxin Alpha Are Ligands for Herpesvirus Entry Mediator. Immunity 1998, 8, 21–30. [Google Scholar] [CrossRef]
- De Togni, P.; Goellner, J.; Ruddle, N.H.; Streeter, P.R.; Fick, A.; Mariathasan, S.; Smith, S.C.; Carlson, R.; Shornick, L.P.; Strauss-Schoenberger, J. Abnormal Development of Peripheral Lymphoid Organs in Mice Deficient in Lymphotoxin. Science 1994, 264, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Koni, P.A.; Sacca, R.; Lawton, P.; Browning, J.L.; Ruddle, N.H.; Flavell, R.A. Distinct Roles in Lymphoid Organogenesis for Lymphotoxins Alpha and Beta Revealed in Lymphotoxin Beta-Deficient Mice. Immunity 1997, 6, 491–500. [Google Scholar] [CrossRef]
- Matsumoto, M.; Mariathasan, S.; Nahm, M.H.; Baranyay, F.; Peschon, J.J.; Chaplin, D.D. Role of Lymphotoxin and the Type I TNF Receptor in the Formation of Germinal Centers. Science 1996, 271, 1289–1291. [Google Scholar] [CrossRef]
- Lin, X.; Ma, X.; Rodriguez, M.; Feng, X.; Zoecklein, L.; Fu, Y.-X.; Roos, R.P. Membrane Lymphotoxin Is Required for Resistance to Theiler’s Virus Infection. Int. Immunol. 2003, 15, 955–962. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Banks, T.A.; Rickert, S.; Benedict, C.A.; Ma, L.; Ko, M.; Meier, J.; Ha, W.; Schneider, K.; Granger, S.W.; Turovskaya, O.; et al. A Lymphotoxin-IFN-Beta Axis Essential for Lymphocyte Survival Revealed during Cytomegalovirus Infection. J. Immunol. Baltim. Md 1950 2005, 174, 7217–7225. [Google Scholar] [CrossRef]
- Guidotti, L.G.; Ishikawa, T.; Hobbs, M.V.; Matzke, B.; Schreiber, R.; Chisari, F.V. Intracellular Inactivation of the Hepatitis B Virus by Cytotoxic T Lymphocytes. Immunity 1996, 4, 25–36. [Google Scholar] [CrossRef]
- Pavić, I.; Polić, B.; Crnković, I.; Lucin, P.; Jonjić, S.; Koszinowski, U.H. Participation of Endogenous Tumour Necrosis Factor Alpha in Host Resistance to Cytomegalovirus Infection. J. Gen. Virol. 1993, 74 (Pt 10), 2215–2223. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ruby, J.; Bluethmann, H.; Peschon, J.J. Antiviral Activity of Tumor Necrosis Factor (TNF) Is Mediated via P55 and P75 TNF Receptors. J. Exp. Med. 1997, 186, 1591–1596. [Google Scholar] [CrossRef]
- Atrasheuskaya, A.V.; Bukin, E.K.; Fredeking, T.M.; Ignatyev, G.M. Protective Effect of Exogenous Recombinant Mouse Interferon-Gamma and Tumour Necrosis Factor-Alpha on Ectromelia Virus Infection in Susceptible BALB/c Mice. Clin. Exp. Immunol. 2004, 136, 207–214. [Google Scholar] [CrossRef]
- Chan, F.K.-M.; Shisler, J.; Bixby, J.G.; Felices, M.; Zheng, L.; Appel, M.; Orenstein, J.; Moss, B.; Lenardo, M.J. A Role for Tumor Necrosis Factor Receptor-2 and Receptor-Interacting Protein in Programmed Necrosis and Antiviral Responses. J. Biol. Chem. 2003, 278, 51613–51621. [Google Scholar] [CrossRef] [PubMed]
- Lidbury, B.A.; Ramshaw, I.A.; Sambhi, S.K. The Role for Host-Immune Factors in the in Vivo Antiviral Effects of Tumour Necrosis Factor. Cytokine 1995, 7, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Sambhi, S.K.; Kohonen-Corish, M.R.; Ramshaw, I.A. Local Production of Tumor Necrosis Factor Encoded by Recombinant Vaccinia Virus Is Effective in Controlling Viral Replication in Vivo. Proc. Natl. Acad. Sci. USA 1991, 88, 4025–4029. [Google Scholar] [CrossRef]
- Alcami, A. Viral Mimicry of Cytokines, Chemokines and Their Receptors. Nat. Rev. Immunol. 2003, 3, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Alejo, A.; Ruiz-Argüello, M.B.; Ho, Y.; Smith, V.P.; Saraiva, M.; Alcami, A. A Chemokine-Binding Domain in the Tumor Necrosis Factor Receptor from Variola (Smallpox) Virus. Proc. Natl. Acad. Sci. USA 2006, 103, 5995–6000. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, G.J.; Buller, R.M.; Glasgow, W.C. Multigenic Evasion of Inflammation by Poxviruses. J. Virol. 1994, 68, 1737–1749. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Hu, F.Q.; Smith, T.D.; Richards, C.L.; Smolak, P.; Goodwin, R.G.; Pickup, D.J. Cowpox Virus Genome Encodes a Second Soluble Homologue of Cellular TNF Receptors, Distinct from CrmB, That Binds TNF but Not LT Alpha. Virology 1996, 223, 132–147. [Google Scholar] [CrossRef]
- Pontejo, S.M.; Alejo, A.; Alcami, A. Comparative Biochemical and Functional Analysis of Viral and Human Secreted Tumor Necrosis Factor (TNF) Decoy Receptors. J. Biol. Chem. 2015, 290, 15973–15984. [Google Scholar] [CrossRef] [PubMed]
- Loparev, V.N.; Parsons, J.M.; Knight, J.C.; Panus, J.F.; Ray, C.A.; Buller, R.M.; Pickup, D.J.; Esposito, J.J. A Third Distinct Tumor Necrosis Factor Receptor of Orthopoxviruses. Proc. Natl. Acad. Sci. USA 1998, 95, 3786–3791. [Google Scholar] [CrossRef] [PubMed]
- Meusch, U.; Rossol, M.; Baerwald, C.; Hauschildt, S.; Wagner, U. Outside-to-inside Signaling through Transmembrane Tumor Necrosis Factor Reverses Pathologic Interleukin-1beta Production and Deficient Apoptosis of Rheumatoid Arthritis Monocytes. Arthritis Rheum. 2009, 60, 2612–2621. [Google Scholar] [CrossRef] [PubMed]
- Al Rumaih, Z.; Tuazon Kels, M.J.; Ng, E.; Pandey, P.; Pontejo, S.M.; Alejo, A.; Alcamí, A.; Chaudhri, G.; Karupiah, G. Poxvirus-Encoded TNF Receptor Homolog Dampens Inflammation and Protects from Uncontrolled Lung Pathology during Respiratory Infection. Proc. Natl. Acad. Sci. USA 2020, 117, 26885–26894. [Google Scholar] [CrossRef] [PubMed]
- Juhász, K.; Buzás, K.; Duda, E. Importance of Reverse Signaling of the TNF Superfamily in Immune Regulation. Expert Rev. Clin. Immunol. 2013, 9, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.C.; Bahar, M.W.; Abrescia, N.G.A.; Smith, G.L.; Stuart, D.I.; Grimes, J.M. Structure of CrmE, a Virus-Encoded Tumour Necrosis Factor Receptor. J. Mol. Biol. 2007, 372, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Upton, C.; Macen, J.L.; Schreiber, M.; McFadden, G. Myxoma Virus Expresses a Secreted Protein with Homology to the Tumor Necrosis Factor Receptor Gene Family That Contributes to Viral Virulence. Virology 1991, 184, 370–382. [Google Scholar] [CrossRef]
- Macen, J.L.; Graham, K.A.; Lee, S.F.; Schreiber, M.; Boshkov, L.K.; McFadden, G. Expression of the Myxoma Virus Tumor Necrosis Factor Receptor Homologue and M11L Genes Is Required to Prevent Virus-Induced Apoptosis in Infected Rabbit T Lymphocytes. Virology 1996, 218, 232–237. [Google Scholar] [CrossRef]
- Sedger, L.; McFadden, G. M-T2: A Poxvirus TNF Receptor Homologue with Dual Activities. Immunol. Cell Biol. 1996, 74, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Sedger, L.M.; Osvath, S.R.; Xu, X.-M.; Li, G.; Chan, F.K.-M.; Barrett, J.W.; McFadden, G. Poxvirus Tumor Necrosis Factor Receptor (TNFR)-like T2 Proteins Contain a Conserved Preligand Assembly Domain That Inhibits Cellular TNFR1-Induced Cell Death. J. Virol. 2006, 80, 9300–9309. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, M.; Smith, P.; Fallon, P.G.; Alcami, A. Inhibition of Type 1 Cytokine-Mediated Inflammation by a Soluble CD30 Homologue Encoded by Ectromelia (Mousepox) Virus. J. Exp. Med. 2002, 196, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Jeng, D.; Singh, R.; Coughlin, J.; Essani, K.; McFadden, G. Interaction of Human TNF and Beta2-Microglobulin with Tanapox Virus-Encoded TNF Inhibitor, TPV-2L. Virology 2009, 386, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Barrett, J.W.; Brouckaert, P.; McFadden, G. Variation in Ligand Binding Specificities of a Novel Class of Poxvirus-Encoded Tumor Necrosis Factor-Binding Protein. J. Biol. Chem. 2006, 281, 22517–22526. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; West, A.P.; Bjorkman, P.J. Crystal Structure of TNFalpha Complexed with a Poxvirus MHC-Related TNF Binding Protein. Nat. Struct. Mol. Biol. 2009, 16, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, J.-L.; Schneider, P.; Tschopp, J. The Molecular Architecture of the TNF Superfamily. Trends Biochem. Sci. 2002, 27, 19–26. [Google Scholar] [CrossRef]
- Hymowitz, S.G.; Christinger, H.W.; Fuh, G.; Ultsch, M.; O’Connell, M.; Kelley, R.F.; Ashkenazi, A.; de Vos, A.M. Triggering Cell Death: The Crystal Structure of Apo2L/TRAIL in a Complex with Death Receptor 5. Mol. Cell 1999, 4, 563–571. [Google Scholar] [CrossRef]
- Pontejo, S.M.; Sanchez, C.; Ruiz-Argüello, B.; Alcami, A. Insights into Ligand Binding by a Viral Tumor Necrosis Factor (TNF) Decoy Receptor Yield a Selective Soluble Human Type 2 TNF Receptor. J. Biol. Chem. 2019, 294, 5214–5227. [Google Scholar] [CrossRef] [PubMed]
- Magis, C.; van der Sloot, A.M.; Serrano, L.; Notredame, C. An Improved Understanding of TNFL/TNFR Interactions Using Structure-Based Classifications. Trends Biochem. Sci. 2012, 37, 353–363. [Google Scholar] [CrossRef]
- Chan, F.K.; Chun, H.J.; Zheng, L.; Siegel, R.M.; Bui, K.L.; Lenardo, M.J. A Domain in TNF Receptors That Mediates Ligand-Independent Receptor Assembly and Signaling. Science 2000, 288, 2351–2354. [Google Scholar] [CrossRef] [PubMed]
- Mukai, Y.; Nakamura, T.; Yoshikawa, M.; Yoshioka, Y.; Tsunoda, S.; Nakagawa, S.; Yamagata, Y.; Tsutsumi, Y. Solution of the Structure of the TNF-TNFR2 Complex. Sci. Signal. 2010, 3, ra83. [Google Scholar] [CrossRef]
- Siegel, R.M.; Frederiksen, J.K.; Zacharias, D.A.; Chan, F.K.; Johnson, M.; Lynch, D.; Tsien, R.Y.; Lenardo, M.J. Fas Preassociation Required for Apoptosis Signaling and Dominant Inhibition by Pathogenic Mutations. Science 2000, 288, 2354–2357. [Google Scholar] [CrossRef]
- Hu, F.Q.; Smith, C.A.; Pickup, D.J. Cowpox Virus Contains Two Copies of an Early Gene Encoding a Soluble Secreted Form of the Type II TNF Receptor. Virology 1994, 204, 343–356. [Google Scholar] [CrossRef]
- Reading, P.C.; Khanna, A.; Smith, G.L. Vaccinia Virus CrmE Encodes a Soluble and Cell Surface Tumor Necrosis Factor Receptor That Contributes to Virus Virulence. Virology 2002, 292, 285–298. [Google Scholar] [CrossRef]
- Saraiva, M.; Alcami, A. CrmE, a Novel Soluble Tumor Necrosis Factor Receptor Encoded by Poxviruses. J. Virol. 2001, 75, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Crowe, P.D.; VanArsdale, T.L.; Walter, B.N.; Ware, C.F.; Hession, C.; Ehrenfels, B.; Browning, J.L.; Din, W.S.; Goodwin, R.G.; Smith, C.A. A Lymphotoxin-Beta-Specific Receptor. Science 1994, 264, 707–710. [Google Scholar] [CrossRef]
- Fenner, F. Smallpox: Emergence, Global Spread, and Eradication. Hist. Philos. Life Sci. 1993, 15, 397–420. [Google Scholar] [PubMed]
- Mavian, C.; López-Bueno, A.; Bryant, N.A.; Seeger, K.; Quail, M.A.; Harris, D.; Barrell, B.; Alcami, A. The Genome Sequence of Ectromelia Virus Naval and Cornell Isolates from Outbreaks in North America. Virology 2014, 462–463, 218–226. [Google Scholar] [CrossRef]
- Ribas, G.; Rivera, J.; Saraiva, M.; Campbell, R.D.; Alcami, A. Genetic Variability of Immunomodulatory Genes in Ectromelia Virus Isolates Detected by Denaturing High-Performance Liquid Chromatography. J. Virol. 2003, 77, 10139–10146. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Alzhanova, D.; Früh, K. Modulation of the Host Immune Response by Cowpox Virus. Microbes Infect. 2010, 12, 900–909. [Google Scholar] [CrossRef] [PubMed]
- Cunnion, K.M. Tumor Necrosis Factor Receptors Encoded by Poxviruses. Mol. Genet. Metab. 1999, 67, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Goebel, S.J.; Johnson, G.P.; Perkus, M.E.; Davis, S.W.; Winslow, J.P.; Paoletti, E. The Complete DNA Sequence of Vaccinia Virus. Virology 1990, 179, 247–266. [Google Scholar] [CrossRef]
- Howard, S.T.; Chan, Y.S.; Smith, G.L. Vaccinia Virus Homologues of the Shope Fibroma Virus Inverted Terminal Repeat Proteins and a Discontinuous ORF Related to the Tumor Necrosis Factor Receptor Family. Virology 1991, 180, 633–647. [Google Scholar] [CrossRef]
- Alcamí, A.; Khanna, A.; Paul, N.L.; Smith, G.L. Vaccinia Virus Strains Lister, USSR and Evans Express Soluble and Cell-Surface Tumour Necrosis Factor Receptors. J. Gen. Virol. 1999, 80 (Pt 4), 949–959. [Google Scholar] [CrossRef] [PubMed]
- Gileva, I.P.; Nepomnyashchikh, T.S.; Antonets, D.V.; Lebedev, L.R.; Kochneva, G.V.; Grazhdantseva, A.V.; Shchelkunov, S.N. Properties of the Recombinant TNF-Binding Proteins from Variola, Monkeypox, and Cowpox Viruses Are Different. Biochim. Biophys. Acta 2006, 1764, 1710–1718. [Google Scholar] [CrossRef]
- Bossen, C.; Ingold, K.; Tardivel, A.; Bodmer, J.-L.; Gaide, O.; Hertig, S.; Ambrose, C.; Tschopp, J.; Schneider, P. Interactions of Tumor Necrosis Factor (TNF) and TNF Receptor Family Members in the Mouse and Human. J. Biol. Chem. 2006, 281, 13964–13971. [Google Scholar] [CrossRef]
- Pontejo, S.M.; Alejo, A.; Alcami, A. Poxvirus-Encoded TNF Decoy Receptors Inhibit the Biological Activity of Transmembrane TNF. J. Gen. Virol. 2015, 96, 3118–3123. [Google Scholar] [CrossRef]
- Kirchner, S.; Boldt, S.; Kolch, W.; Haffner, S.; Kazak, S.; Janosch, P.; Holler, E.; Andreesen, R.; Eissner, G. LPS Resistance in Monocytic Cells Caused by Reverse Signaling through Transmembrane TNF (MTNF) Is Mediated by the MAPK/ERK Pathway. J. Leukoc. Biol. 2004, 75, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Alejo, A.; Ruiz-Argüello, M.B.; Pontejo, S.M.; Fernández de Marco, M.D.M.; Saraiva, M.; Hernáez, B.; Alcamí, A. Chemokines Cooperate with TNF to Provide Protective Anti-Viral Immunity and to Enhance Inflammation. Nat. Commun. 2018, 9, 1790. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Lu, Q.; Wei, H.; Wang, D.; Chen, D.; He, G.; Huang, L.; Wang, H.; Wang, X. Structural Basis of Chemokine Sequestration by CrmD, a Poxvirus-Encoded Tumor Necrosis Factor Receptor. PLoS Pathog. 2011, 7, e1002162. [Google Scholar] [CrossRef] [PubMed]
- Heidarieh, H.; Hernáez, B.; Alcamí, A. Immune Modulation by Virus-Encoded Secreted Chemokine Binding Proteins. Virus Res. 2015, 209, 67–75. [Google Scholar] [CrossRef]
- Smith, G.L.; Benfield, C.T.O.; Maluquer de Motes, C.; Mazzon, M.; Ember, S.W.J.; Ferguson, B.J.; Sumner, R.P. Vaccinia Virus Immune Evasion: Mechanisms, Virulence and Immunogenicity. J. Gen. Virol. 2013, 94, 2367–2392. [Google Scholar] [CrossRef] [PubMed]
- Felix, J.; Savvides, S.N. Mechanisms of Immunomodulation by Mammalian and Viral Decoy Receptors: Insights from Structures. Nat. Rev. Immunol. 2017, 17, 112–129. [Google Scholar] [CrossRef]
- Nelson, C.A.; Epperson, M.L.; Singh, S.; Elliott, J.I.; Fremont, D.H. Structural Conservation and Functional Diversity of the Poxvirus Immune Evasion (PIE) Domain Superfamily. Viruses 2015, 7, 4878–4898. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Davis, T.; Anderson, D.; Solam, L.; Beckmann, M.P.; Jerzy, R.; Dower, S.K.; Cosman, D.; Goodwin, R.G. A Receptor for Tumor Necrosis Factor Defines an Unusual Family of Cellular and Viral Proteins. Science 1990, 248, 1019–1023. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, M.; McFadden, G. Mutational Analysis of the Ligand-Binding Domain of M-T2 Protein, the Tumor Necrosis Factor Receptor Homologue of Myxoma Virus. J. Immunol. 1996, 157, 4486–4495. [Google Scholar] [PubMed]
- Smith, C.A.; Davis, T.; Wignall, J.M.; Din, W.S.; Farrah, T.; Upton, C.; McFadden, G.; Goodwin, R.G. T2 Open Reading Frame from the Shope Fibroma Virus Encodes a Soluble Form of the TNF Receptor. Biochem. Biophys. Res. Commun. 1991, 176, 335–342. [Google Scholar] [CrossRef]
- Schreiber, M.; Sedger, L.; McFadden, G. Distinct Domains of M-T2, the Myxoma Virus Tumor Necrosis Factor (TNF) Receptor Homolog, Mediate Extracellular TNF Binding and Intracellular Apoptosis Inhibition. J. Virol. 1997, 71, 2171–2181. [Google Scholar] [CrossRef]
- McFadden, G.; Schreiber, M.; Sedger, L. Myxoma T2 Protein as a Model for Poxvirus TNF Receptor Homologs. J. Neuroimmunol. 1997, 72, 119–126. [Google Scholar] [CrossRef]
- Horie, R.; Watanabe, T. CD30: Expression and Function in Health and Disease. Semin. Immunol. 1998, 10, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Panus, J.F.; Smith, C.A.; Ray, C.A.; Smith, T.D.; Patel, D.D.; Pickup, D.J. Cowpox Virus Encodes a Fifth Member of the Tumor Necrosis Factor Receptor Family: A Soluble, Secreted CD30 Homologue. Proc. Natl. Acad. Sci. USA 2002, 99, 8348–8353. [Google Scholar] [CrossRef] [PubMed]
- Afonso, C.L.; Delhon, G.; Tulman, E.R.; Lu, Z.; Zsak, A.; Becerra, V.M.; Zsak, L.; Kutish, G.F.; Rock, D.L. Genome of Deerpox Virus. J. Virol. 2005, 79, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Alejo, A.; Saraiva, M.; Ruiz-Argüello, M.B.; Viejo-Borbolla, A.; de Marco, M.F.; Salguero, F.J.; Alcami, A. A Method for the Generation of Ectromelia Virus (ECTV) Recombinants: In Vivo Analysis of ECTV VCD30 Deletion Mutants. PLoS ONE 2009, 4, e5175. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, C.R.; Paulose-Murphy, M.; Singh, R.; Qin, J.; Barrett, J.W.; Tardivel, A.; Schneider, P.; Essani, K.; McFadden, G. A Secreted High-Affinity Inhibitor of Human TNF from Tanapox Virus. Proc. Natl. Acad. Sci. USA 2003, 100, 4831–4836. [Google Scholar] [CrossRef] [PubMed]
- Chapman, T.L.; Bjorkman, P.J. Characterization of a Murine Cytomegalovirus Class I Major Histocompatibility Complex (MHC) Homolog: Comparison to MHC Molecules and to the Human Cytomegalovirus MHC Homolog. J. Virol. 1998, 72, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Farrell, H.E.; Vally, H.; Lynch, D.M.; Fleming, P.; Shellam, G.R.; Scalzo, A.A.; Davis-Poynter, N.J. Inhibition of Natural Killer Cells by a Cytomegalovirus MHC Class I Homologue in Vivo. Nature 1997, 386, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Senkevich, T.G.; Moss, B. Domain Structure, Intracellular Trafficking, and Beta2-Microglobulin Binding of a Major Histocompatibility Complex Class I Homolog Encoded by Molluscum Contagiosum Virus. Virology 1998, 250, 397–407. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Afonso, C.L.; Tulman, E.R.; Lu, Z.; Zsak, L.; Osorio, F.A.; Balinsky, C.; Kutish, G.F.; Rock, D.L. The Genome of Swinepox Virus. J. Virol. 2002, 76, 783–790. [Google Scholar] [CrossRef]
- Wong, M.; Ziring, D.; Korin, Y.; Desai, S.; Kim, S.; Lin, J.; Gjertson, D.; Braun, J.; Reed, E.; Singh, R.R. TNFalpha Blockade in Human Diseases: Mechanisms and Future Directions. Clin. Immunol. 2008, 126, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P.; Kalden, J.R. Advances in Rheumatology: New Targeted Therapeutics. Arthritis Res. Ther. 2011, 13 (Suppl. 1), S5. [Google Scholar] [CrossRef]
- Mitoma, H.; Horiuchi, T.; Tsukamoto, H.; Ueda, N. Molecular Mechanisms of Action of Anti-TNF-α Agents - Comparison among Therapeutic TNF-α Antagonists. Cytokine 2018, 101, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Tsiodras, S.; Samonis, G.; Boumpas, D.T.; Kontoyiannis, D.P. Fungal Infections Complicating Tumor Necrosis Factor Alpha Blockade Therapy. Mayo Clin. Proc. 2008, 83, 181–194. [Google Scholar] [CrossRef]
- Kim, E.-M.; Uhm, W.-S.; Bae, S.-C.; Yoo, D.-H.; Kim, T.-H. Incidence of Tuberculosis among Korean Patients with Ankylosing Spondylitis Who Are Taking Tumor Necrosis Factor Blockers. J. Rheumatol. 2011, 38, 2218–2223. [Google Scholar] [CrossRef]
- Yaron, J.R.; Zhang, L.; Guo, Q.; Burgin, M.; Schutz, L.N.; Awo, E.; Wise, L.; Krause, K.L.; Ildefonso, C.J.; Kwiecien, J.M.; et al. Deriving Immune Modulating Drugs from Viruses—A New Class of Biologics. J. Clin. Med. 2020, 9, 972. [Google Scholar] [CrossRef]
- Gileva, I.P.; Viazovaia, E.A.; Toporkova, L.B.; Tsyrendorzhiev, D.D.; Shchelkunov, S.N.; Orlovskaya, I.A. TNF Binding Protein of Variola Virus Acts as a TNF Antagonist at Epicutaneous Application. Curr. Pharm. Biotechnol. 2015, 16, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Viazovaia, E.A.; Gileva, I.P.; Toporkova, L.B.; Shchelkunov, S.N.; Orlovskaya, I.A. Anti-Inflammatory Effects of Variola Virus TNF Decoy Receptor in an Experimental Model of Contact Dermatitis. Curr. Pharm. Biotechnol. 2018, 19, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Shchelkunov, S.N.; Taranov, O.S.; Tregubchak, T.V.; Maksyutov, R.A.; Silkov, A.N.; Nesterov, A.E.; Sennikov, S.V. The Gene Therapy of Collagen-Induced Arthritis in Rats by Intramuscular Administration of the Plasmid Encoding TNF-Binding Domain of Variola Virus CrmB Protein. Dokl. Biochem. Biophys. 2016, 469, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Hopkin, S.J.; Lewis, J.W.; Krautter, F.; Chimen, M.; McGettrick, H.M. Triggering the Resolution of Immune Mediated Inflammatory Diseases: Can Targeting Leukocyte Migration Be the Answer? Front. Pharmacol. 2019, 10, 184. [Google Scholar] [CrossRef] [PubMed]
- Alejo, A.; Sánchez, C.; Amu, S.; Fallon, P.G.; Alcamí, A. Addition of a Viral Immunomodulatory Domain to Etanercept Generates a Bifunctional Chemokine and TNF Inhibitor. J. Clin. Med. 2019, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Kruglov, A.A.; Grivennikov, S.I.; Kuprash, D.V.; Winsauer, C.; Prepens, S.; Seleznik, G.M.; Eberl, G.; Littman, D.R.; Heikenwalder, M.; Tumanov, A.V.; et al. Nonredundant Function of Soluble LTα3 Produced by Innate Lymphoid Cells in Intestinal Homeostasis. Science 2013, 342, 1243–1246. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, V.; Fu, Y.-X. Lymphotoxin Signalling in Immune Homeostasis and the Control of Microorganisms. Nat. Rev. Immunol. 2013, 13, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.-H.; Cohen, M.; Tang, Y.; Lazear, E.; Whitbeck, J.C.; Eisenberg, R.J.; Cohen, G.H.; Sigal, L.J. The Orthopoxvirus Type I IFN Binding Protein Is Essential for Virulence and an Effective Target for Vaccination. J. Exp. Med. 2008, 205, 981–992. [Google Scholar] [CrossRef] [PubMed]
- Antoine, G.; Scheiflinger, F.; Dorner, F.; Falkner, F.G. The Complete Genomic Sequence of the Modified Vaccinia Ankara Strain: Comparison with Other Orthopoxviruses. Virology 1998, 244, 365–396. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, T.J.; Alcami, A.; Andrea, P.; Smith, G.L. Modified Vaccinia Virus Ankara Undergoes Limited Replication in Human Cells and Lacks Several Immunomodulatory Proteins: Implications for Use as a Human Vaccine. J. Gen. Virol. 1998, 79 (Pt 5), 1159–1167. [Google Scholar] [CrossRef]
- Alcamí, A.; Symons, J.A.; Smith, G.L. The Vaccinia Virus Soluble Alpha/Beta Interferon (IFN) Receptor Binds to the Cell Surface and Protects Cells from the Antiviral Effects of IFN. J. Virol. 2000, 74, 11230–11239. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Protein | Key Refs. | Virus | Virulence Factor | Known Ligands | Cellular Homology | kDa | Special Traits | |
|---|---|---|---|---|---|---|---|---|
| vTNFRs | CrmB | [33,34] | CMPXV, CPXV, MPXV, VARV | Unclear (~50-fold LD50 reduction after intracranial inoculation) | hTNF, mTNF, hLTα, mLTα | TNFR2 | 48 | SECRET domain-mediated chemokine inhibition; Reverse signalling |
| CrmC | [35,36] | CPXV, VACV | Minor effect (difference in weight loss) | hTNF, mTNF | TNFR2 | 25 | Reverse signalling | |
| CrmD | [37,38,39,40] | CPXV, ECTV | Yes (reduced LD50 in 6 orders of magnitude) | hTNF, mTNF, hLTα, mLTα | TNFR2 | 46 | SECRET domain-mediated chemokine inhibition; Reverse signalling | |
| CrmE | [41] | CPXV, VACV | Minor effect (difference in weight loss) | hTNF, mTNF | TNFR2 | 18 | Mouse tmTNF inhibition w/o sTNF; Reverse signalling | |
| T2 | [42,43,44,45] | MYXV | Yes (reduce mortality) | rabbit TNF | TNFR2 | M-T2: 40.5 | PLAD mediated antiapoptotic activity | |
| SFV | hTNF, rabbit TNF | TNFR2 | S-T2: 58 | |||||
| vCD30 | [46] | DPXV, CPXV, ECTV | No | CD30L | CD30 | 12 | Reverse signalling through CD30L; inhibition of IFNγ production in splenocytes | |
| vTNFBPs | 2L | [47,48,49] | TPXV | ND | rabbit, human, monkey, canine TNF | MHC I heavy chain | 47 | Can associate with β2 microglobulin to inhibit MHC-I |
| YMTV | rabbit, human, monkey TNF | |||||||
| SPV003 | [48] | SPXV | ND | porcine TNF | MHC I heavy chain | 47 | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alvarez-de Miranda, F.J.; Alonso-Sánchez, I.; Alcamí, A.; Hernaez, B. TNF Decoy Receptors Encoded by Poxviruses. Pathogens 2021, 10, 1065. https://doi.org/10.3390/pathogens10081065
Alvarez-de Miranda FJ, Alonso-Sánchez I, Alcamí A, Hernaez B. TNF Decoy Receptors Encoded by Poxviruses. Pathogens. 2021; 10(8):1065. https://doi.org/10.3390/pathogens10081065
Chicago/Turabian StyleAlvarez-de Miranda, Francisco Javier, Isabel Alonso-Sánchez, Antonio Alcamí, and Bruno Hernaez. 2021. "TNF Decoy Receptors Encoded by Poxviruses" Pathogens 10, no. 8: 1065. https://doi.org/10.3390/pathogens10081065
APA StyleAlvarez-de Miranda, F. J., Alonso-Sánchez, I., Alcamí, A., & Hernaez, B. (2021). TNF Decoy Receptors Encoded by Poxviruses. Pathogens, 10(8), 1065. https://doi.org/10.3390/pathogens10081065