
Completing the Genome Sequence of Chlamydia pecorum Strains MC/MarsBar and DBDeUG: New Insights into This Enigmatic Koala (Phascolarctos cinereus) Pathogen

 ,
,  , , and
, , and
Abstract
1. Introduction
2. Results
2.1. High Quality Closed Genomes of C. pecorum DBDeUG_2018 and MC/MarsBar_2018
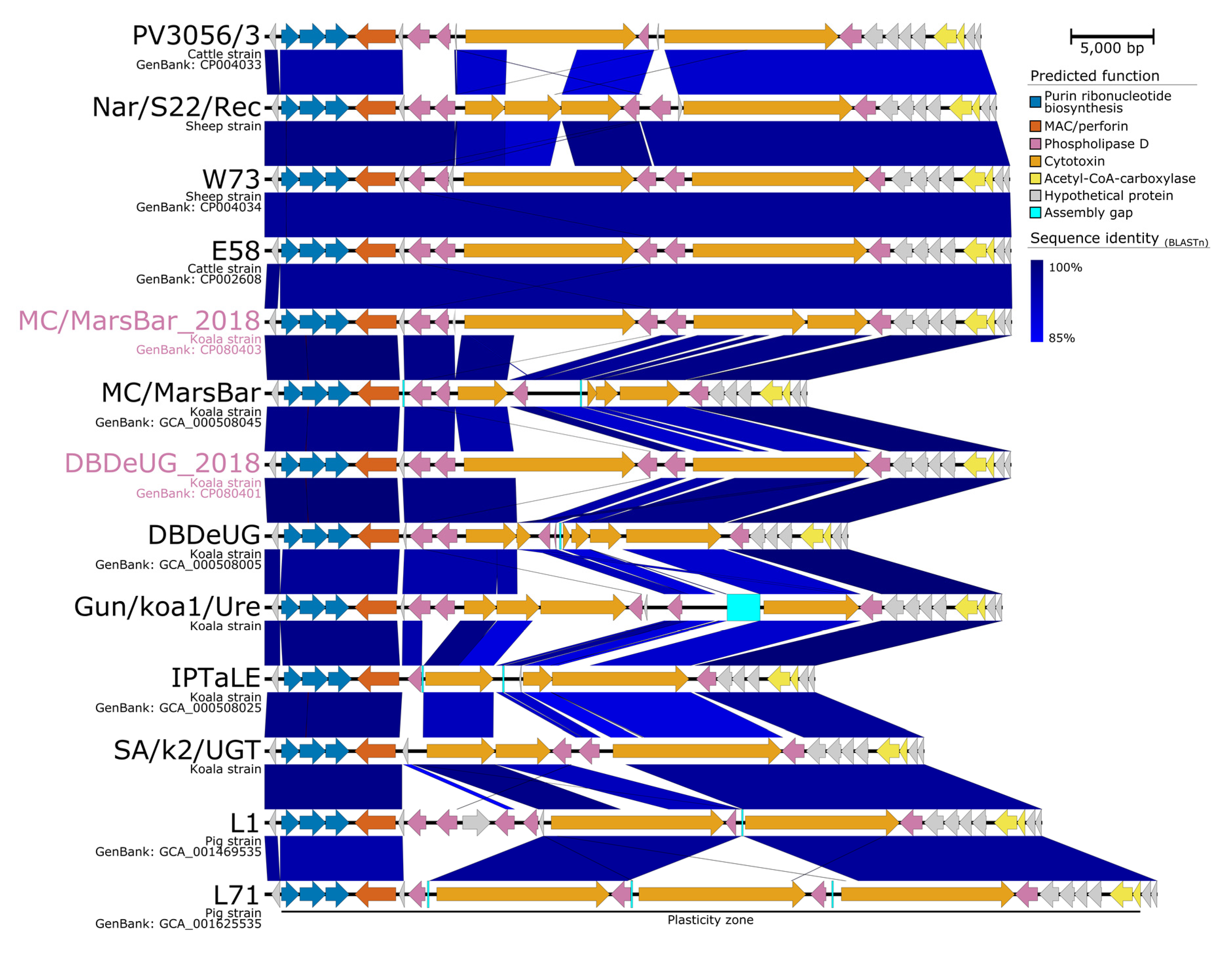
2.2. Improved Annotations of C. pecorum Regions of Interest
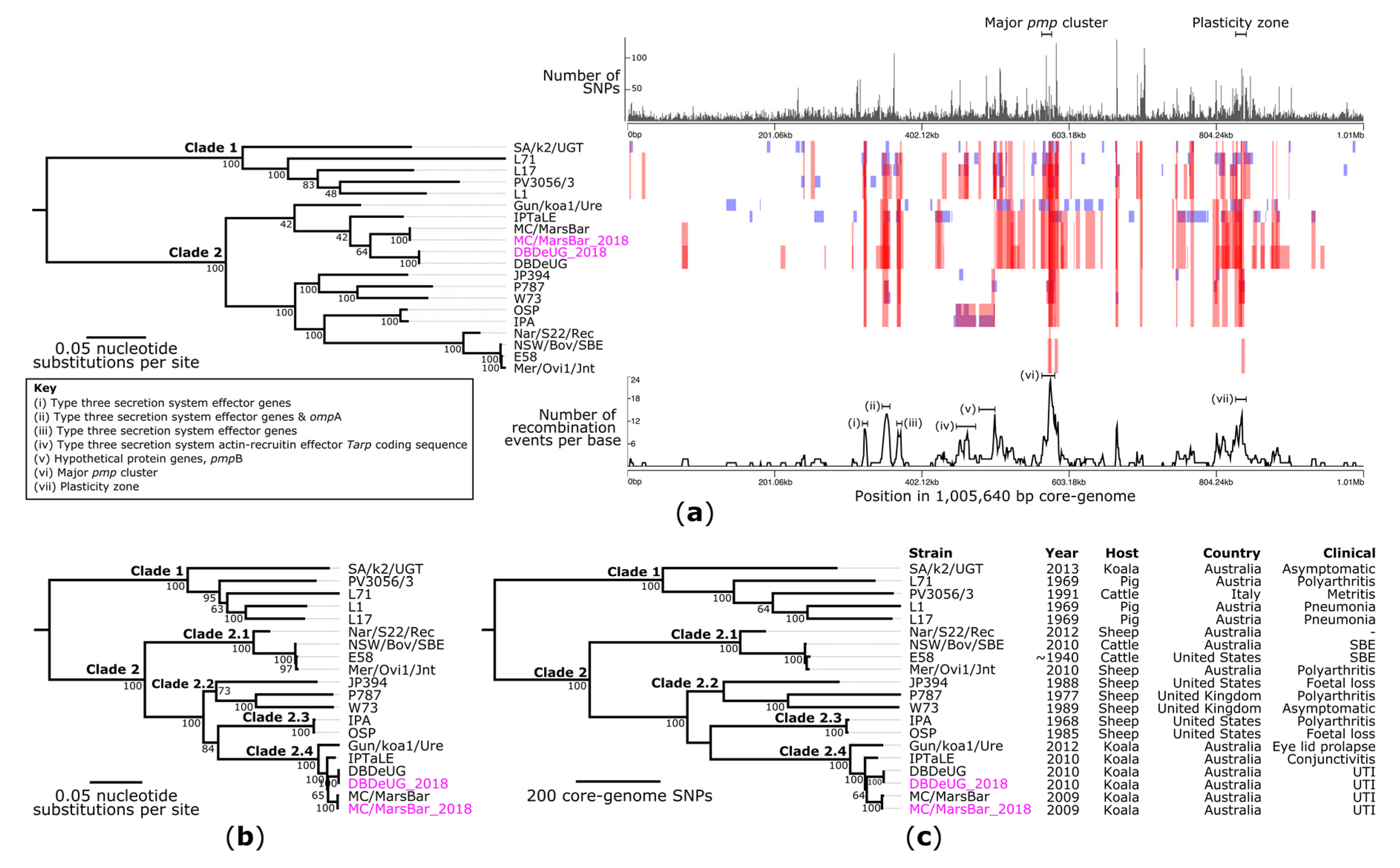
2.3. Phylogenetic Analyses of C. pecorum Confirm Separation of Livestock from Koala Strains
2.4. Comparative Genomics with Other Koala-Derived C. pecorum Reveals New Loci of Interest Accumulating SNPs
3. Discussion
3.1. Koala C. pecorum Genomics Revisited
3.2. Our Effort to Broaden Understanding of C. pecorum Virulence Factors
3.3. Phylogenetic and Comparative Genomic Analyses of Koala C. pecorum Identify New Genetic Markers That Could Be Used for Intra-Species Fine-Detailed Molecular Epidemiology
3.4. Need for Further Research into the Role of Chlamydial Plasmids in C. pecorum Infections
4. Materials and Methods
4.1. Chlamydia Pecorum Sample Description, Genome, Plasmid Copy Number Quantification, and Genome Sequencing
4.2. Quality Control of Sequence Data
4.3. De Novo Genome Assembly, Genome Analyses, and In Silico Multilocus Sequence Typing
4.4. Variant Detection and Phylogenetic Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Borel, N.; Polkinghorne, A.; Pospischil, A. A review on chlamydial diseases in animals: Still a challenge for pathologists? Vet. Pathol. 2018, 55, 374–390. [Google Scholar] [CrossRef]
- Quigley, B.L.; Timms, P. Helping koalas battle disease-Recent advances in Chlamydia and koala retrovirus (KoRV) disease understanding and treatment in koalas. FEMS Microbiol. Rev. 2020, 44, 583–605. [Google Scholar] [CrossRef]
- Griffith, J.E.; Dhand, N.K.; Krockenberger, M.B.; Higgins, D.P. A retrospective study of admission trends of koalas to a rehabilitation facility over 30 years. J. Wildl. Dis. 2013, 49, 18–28. [Google Scholar] [CrossRef]
- Gonzalez-Astudillo, V.; Allavena, R.; McKinnon, A.; Larkin, R.; Henning, J. Decline causes of Koalas in South East Queensland, Australia: A 17-year retrospective study of mortality and morbidity. Sci. Rep. 2017, 7, 42587. [Google Scholar] [CrossRef]
- Legione, A.R.; Patterson, J.L.; Whiteley, P.L.; Amery-Gale, J.; Lynch, M.; Haynes, L.; Gilkerson, J.R.; Polkinghorne, A.; Devlin, J.M.; Sansom, F.M. Identification of unusual Chlamydia pecorum genotypes in Victorian koalas (Phascolarctos cinereus) and clinical variables associated with infection. J. Med. Microbiol. 2016, 65, 420–428. [Google Scholar] [CrossRef]
- Fernandez, C.M.; Schmertmann, L.J.; Higgins, D.P.; Casteriano, A.; Irinyi, L.; Mella, V.S.; Crowther, M.S.; Meyer, W.; Krockenberger, M.B. Genetic differences in Chlamydia pecorum between neighbouring sub-populations of koalas (Phascolarctos cinereus). Vet. Microbiol. 2019, 231, 264–270. [Google Scholar] [CrossRef]
- Robbins, A.; Hanger, J.; Jelocnik, M.; Quigley, B.L.; Timms, P. Longitudinal study of wild koalas (Phascolarctos cinereus) reveals chlamydial disease progression in two thirds of infected animals. Sci. Rep. 2019, 9, 13194. [Google Scholar] [CrossRef]
- Robbins, A.; Hanger, J.; Jelocnik, M.; Quigley, B.L.; Timms, P. Koala immunogenetics and chlamydial strain type are more directly involved in chlamydial disease progression in koalas from two South East Queensland koala populations than koala retrovirus subtypes. Sci. Rep. 2020, 10, 15013. [Google Scholar] [CrossRef]
- Clune, T.; Besier, S.; Hair, S.; Hancock, S.; Lockwood, A.; Thompson, A.; Jelocnik, M.; Jacobson, C. Chlamydia pecorum detection in aborted and stillborn lambs from Western Australia. Vet. Res. 2021, 52, 84. [Google Scholar] [CrossRef]
- Rohner, L.; Marti, H.; Torgerson, P.; Hoffmann, K.; Jelocnik, M.; Borel, N. Prevalence and molecular characterization of C. pecorum detected in Swiss fattening pigs. Vet. Microbiol. 2021, 256, 109062. [Google Scholar] [CrossRef]
- Jelocnik, M. Chlamydiae from down under: The curious cases of chlamydial infections in Australia. Microorganisms 2019, 7, 602. [Google Scholar] [CrossRef]
- Jelocnik, M.; Frentiu, F.D.; Timms, P.; Polkinghorne, A. Multilocus sequence analysis provides insights into molecular epidemiology of Chlamydia pecorum infections in Australian sheep, cattle, and koalas. J. Clin. Microbiol. 2013, 51, 2625–2632. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Heather, J.M.; Chain, B. The sequence of sequencers: The history of sequencing DNA. Genomics 2016, 107, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Branley, J.; Bachmann, N.L.; Jelocnik, M.; Myers, G.S.; Polkinghorne, A. Australian human and parrot Chlamydia psittaci strains cluster within the highly virulent 6BC clade of this important zoonotic pathogen. Sci. Rep. 2016, 6, 30019. [Google Scholar] [CrossRef] [PubMed]
- Hölzer, M.; Barf, L.M.; Lamkiewicz, K.; Vorimore, F.; Lataretu, M.; Favaroni, A.; Schnee, C.; Laroucau, K.; Marz, M.; Sachse, K. Comparative genome analysis of 33 Chlamydia strains reveals characteristic features of Chlamydia psittaci and closely related species. Pathogens 2020, 9, 899. [Google Scholar] [CrossRef]
- Hadfield, J.; Harris, S.R.; Seth-Smith, H.M.; Parmar, S.; Andersson, P.; Giffard, P.M.; Schachter, J.; Moncada, J.; Ellison, L.; Vaulet, M.L.G.; et al. Comprehensive global genome dynamics of Chlamydia trachomatis show ancient diversification followed by contemporary mixing and recent lineage expansion. Genome Res. 2017, 27, 1220–1229. [Google Scholar] [CrossRef]
- Bachmann, N.L.; Fraser, T.A.; Bertelli, C.; Jelocnik, M.; Gillett, A.; Funnell, O.; Flanagan, C.; Myers, G.S.; Timms, P.; Polkinghorne, A. Comparative genomics of koala, cattle and sheep strains of Chlamydia pecorum. BMC Genom. 2014, 15, 667. [Google Scholar] [CrossRef]
- Mojica, S.; Huot Creasy, H.; Daugherty, S.; Read, T.D.; Kim, T.; Kaltenboeck, B.; Bavoil, P.; Myers, G.S. Genome sequence of the obligate intracellular animal pathogen Chlamydia pecorum E58. J. Bacteriol. 2011, 193, 3690. [Google Scholar] [CrossRef]
- Sait, M.; Livingstone, M.; Clark, E.M.; Wheelhouse, N.; Spalding, L.; Markey, B.; Magnino, S.; Lainson, F.A.; Myers, G.S.; Longbottom, D. Genome sequencing and comparative analysis of three Chlamydia pecorum strains associated with different pathogenic outcomes. BMC Genom. 2014, 15, 23. [Google Scholar] [CrossRef]
- Jelocnik, M.; Bachmann, N.L.; Kaltenboeck, B.; Waugh, C.; Woolford, L.; Speight, K.N.; Gillett, A.; Higgins, D.P.; Flanagan, C.; Myers, G.S.; et al. Genetic diversity in the plasticity zone and the presence of the chlamydial plasmid differentiates Chlamydia pecorum strains from pigs, sheep, cattle, and koalas. BMC Genom. 2015, 16, 893. [Google Scholar] [CrossRef]
- Bachmann, N.L.; Sullivan, M.J.; Jelocnik, M.; Myers, G.S.; Timms, P.; Polkinghorne, A. Culture-independent genome sequencing of clinical samples reveals an unexpected heterogeneity of infections by Chlamydia pecorum. J. Clin. Microbiol. 2015, 53, 1573–1581. [Google Scholar] [CrossRef]
- Joseph, S.J.; Marti, H.; Didelot, X.; Castillo-Ramirez, S.; Read, T.D.; Dean, D. Chlamydiaceae genomics reveals interspecies admixture and the recent evolution of Chlamydia abortus infecting lower mammalian species and humans. Genome Biol. Evol. 2015, 7, 3070–3084. [Google Scholar] [CrossRef]
- Bachmann, N.L.; Polkinghorne, A.; Timms, P. Chlamydia genomics: Providing novel insights into chlamydial biology. Trends Microbiol. 2014, 22, 464–472. [Google Scholar] [CrossRef]
- Gorrie, C.L.; Da Silva, A.G.; Ingle, D.J.; Higgs, C.; Seemann, T.; Stinear, T.P.; Williamson, D.A.; Kwong, J.C.; Grayson, M.L.; Sherry, N.L.; et al. Key parameters for genomics-based real-time detection and tracking of multidrug-resistant bacteria: A systematic analysis. Lancet Microbe 2021, 2, e575–e583, in press. [Google Scholar] [CrossRef]
- Taylor-Brown, A.; Madden, D.; Polkinghorne, A. Culture-independent approaches to chlamydial genomics. Microb. Genom. 2018, 4, e000145. [Google Scholar] [CrossRef] [PubMed]
- Zaręba-Marchewka, K.; Szymańska-Czerwińska, M.; Niemczuk, K. Chlamydiae-what’s new? J. Vet. Res. 2020, 64, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Jelocnik, M.; Bachmann, N.L.; Seth-Smith, H.; Thomson, N.R.; Timms, P.; Polkinghorne, A.M. Molecular characterisation of the Chlamydia pecorum plasmid from porcine, ovine, bovine, and koala strains indicates plasmid-strain co-evolution. PeerJ 2016, 4, e1661. [Google Scholar] [CrossRef] [PubMed]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 2018, 3, 124. [Google Scholar] [CrossRef] [PubMed]
- Alikhan, N.F.; Petty, N.K.; Zakour, N.L.B.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple prokaryote genome comparisons. BMC Genom. 2011, 12, 402. [Google Scholar] [CrossRef]
- Sigalova, O.M.; Chaplin, A.V.; Bochkareva, O.O.; Shelyakin, P.V.; Filaretov, V.A.; Akkuratov, E.E.; Burskaia, V.; Gelfand, M.S. Chlamydia pan-genomic analysis reveals balance between host adaptation and selective pressure to genome reduction. BMC Genom. 2019, 20, 710. [Google Scholar] [CrossRef]
- Gomes, J.P.; Bruno, W.J.; Borrego, M.J.; Dean, D. Recombination in the genome of Chlamydia trachomatis involving the polymorphic membrane protein C gene relative to ompA and evidence for horizontal gene transfer. J. Bacteriol. 2004, 186, 4295–4306. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, B.M.; Suchland, R.J.; Quinn, K.L.; Davidson, J.R.; Stamm, W.E.; Rockey, D.D. Genome sequencing of recent clinical Chlamydia trachomatis strains identifies loci associated with tissue tropism and regions of apparent recombination. Infect. Immun. 2010, 78, 2544–2553. [Google Scholar] [CrossRef]
- Joseph, S.J.; Didelot, X.; Gandhi, K.; Dean, D.; Read, T.D. Interplay of recombination and selection in the genomes of Chlamydia trachomatis. Biol. Direct. 2011, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.R.; Clarke, I.N.; Seth-Smith, H.M.; Solomon, A.W.; Cutcliffe, L.T.; Marsh, P.; Skilton, R.J.; Holland, M.J.; Mabey, D.; Peeling, R.W.; et al. Whole-genome analysis of diverse Chlamydia trachomatis strains identifies phylogenetic relationships masked by current clinical typing. Nat. Genet. 2012, 44, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.; Antelo, M.; Nunes, A.; Borges, V.; Damião, V.; Borrego, M.J.; Gomes, J.P. In silico scrutiny of genes revealing phylogenetic congruence with clinical prevalence or tropism properties of Chlamydia trachomatis strains. G3 Genes Genomes Genet. 2014, 5, 9–19. [Google Scholar] [CrossRef]
- Islam, M.M.; Jelocnik, M.; Huston, W.M.; Timms, P.; Polkinghorne, A. Characterization of the in vitro Chlamydia pecorum response to gamma interferon. Infect. Immun. 2018, 86, e00714–e00717. [Google Scholar] [CrossRef]
- Islam, M.M.; Jelocnik, M.; Anstey, S.; Kaltenboeck, B.; Borel, N.; Timms, P.; Polkinghorne, A. In vitro analysis of genetically distinct Chlamydia pecorum isolates reveals key growth differences in mammalian epithelial and immune cells. Vet. Microbiol. 2019, 232, 22–29. [Google Scholar] [CrossRef]
- Bugalhão, J.N.; Mota, L.J. The multiple functions of the numerous Chlamydia trachomatis secreted proteins: The tip of the iceberg. Microb. Cell 2019, 6, 414–449. [Google Scholar] [CrossRef]
- Gitsels, A.; Sanders, N.; Vanrompay, D. Chlamydial infection from outside to inside. Front. Microbiol. 2019, 10, 2329. [Google Scholar] [CrossRef] [PubMed]
- Andersen, S.E.; Bulman, L.M.; Steiert, B.; Faris, R.; Weber, M.M. Got mutants? How advances in chlamydial genetics have furthered the study of effector proteins. Pathog. Dis. 2021, 79, ftaa078. [Google Scholar] [CrossRef]
- Mojica, S.A.; Hovis, K.M.; Frieman, M.B.; Tran, B.; Hsia, R.C.; Ravel, J.; Jenkins-Houk, C.; Wilson, K.L.; Bavoil, P.M. SINC, a type III secreted protein of Chlamydia psittaci, targets the inner nuclear membrane of infected cells and uninfected neighbors. Mol. Biol. Cell 2015, 26, 1918–1934. [Google Scholar] [CrossRef] [PubMed]
- Caven, L.; Carabeo, R.A. Pathogenic puppetry: Manipulation of the host actin cytoskeleton by Chlamydia trachomatis. Int. J. Mol. Sci. 2019, 21, 90. [Google Scholar] [CrossRef] [PubMed]
- Marsh, J.; Kollipara, A.; Timms, P.; Polkinghorne, A. Novel molecular markers of Chlamydia pecorum genetic diversity in the koala (Phascolarctos cinereus). BMC Microbiol. 2011, 11, 77. [Google Scholar] [CrossRef]
- Rajaram, K.; Giebel, A.M.; Toh, E.; Hu, S.; Newman, J.H.; Morrison, S.G.; Kari, L.; Morrison, R.P.; Nelson, D.E. Mutational analysis of the Chlamydia muridarum plasticity zone. Infect. Immun. 2015, 83, 2870–2881. [Google Scholar] [CrossRef] [PubMed]
- Bothe, M.; Dutow, P.; Pich, A.; Genth, H.; Klos, A. DXD motif-dependent and -independent effects of the Chlamydia trachomatis cytotoxin CT166. Toxins 2015, 7, 621–637. [Google Scholar] [CrossRef]
- Klapproth, J.M.A.; Sasaki, M.; Sherman, M.; Babbin, B.; Donnenberg, M.S.; Fernandes, P.J.; Scaletsky, I.C.; Kalman, D.; Nusrat, A.; Williams, I.R. Citrobacter rodentium lifA/efa1 is essential for colonic colonization and crypt cell hyperplasia in vivo. Infect. Immun. 2005, 73, 1441–1451. [Google Scholar] [CrossRef] [PubMed]
- Read, T.D.; Joseph, S.J.; Didelot, X.; Liang, B.; Patel, L.; Dean, D. Comparative analysis of Chlamydia psittaci genomes reveals the recent emergence of a pathogenic lineage with a broad host range. mBio 2013, 4, e00604–e00612. [Google Scholar] [CrossRef] [PubMed]
- Wolff, B.J.; Morrison, S.S.; Pesti, D.; Ganakammal, S.R.; Srinivasamoorthy, G.; Changayil, S.; Weil, M.R.; MacCannell, D.; Rowe, L.; Frace, M.; et al. Chlamydia psittaci comparative genomics reveals intraspecies variations in the putative outer membrane and type III secretion system genes. Microbiology 2015, 161, 1378–1391. [Google Scholar] [CrossRef]
- Ramsey, K.H.; Schripsema, J.H.; Smith, B.J.; Wang, Y.; Jham, B.C.; O’hagan, K.P.; Thomson, N.R.; Murthy, A.K.; Skilton, R.J.; Chu, P.; et al. Plasmid CDS5 influences infectivity and virulence in a mouse model of Chlamydia trachomatis urogenital infection. Infect. Immun. 2014, 82, 3341–3349. [Google Scholar] [CrossRef]
- Shao, L.; Zhang, T.; Melero, J.; Huang, Y.; Liu, Y.; Liu, Q.; He, C.; Nelson, D.E.; Zhong, G. The genital tract virulence factor pGP3 is essential for Chlamydia muridarum colonization in the gastrointestinal tract. Infect. Immun. 2018, 86, e00429-17. [Google Scholar] [CrossRef]
- Jenkins, C.; Jelocnik, M.; Onizawa, E.; McNally, J.; Coilparampil, R.; Pinczowski, P.; Bogema, D.; Westermann, T. Chlamydia pecorum ovine abortion: Associations between maternal infection and perinatal mortality. Pathogens 2021, 10, 1367. [Google Scholar] [CrossRef]
- Lawrence, A.; Fraser, T.; Gillett, A.; Tyndall, J.D.; Timms, P.; Polkinghorne, A.; Huston, W.M. Chlamydia serine protease inhibitor, targeting HtrA, as a new treatment for koala Chlamydia infection. Sci. Rep. 2016, 6, 31466. [Google Scholar] [CrossRef]
- Jelocnik, M.; Laurence, M.; Murdoch, F.R.; Polkinghorne, A. Detection of Chlamydiaceae in ocular swabs from Australian pre-export feedlot sheep. Aust. Vet. J. 2019, 97, 401–403. [Google Scholar] [CrossRef]
- Phillips, S.; Robbins, A.; Loader, J.; Hanger, J.; Booth, R.; Jelocnik, M.; Polkinghorne, A.; Timms, P. Chlamydia pecorum gastrointestinal tract infection associations with urogenital tract infections in the koala (Phascolarctos cinereus). PLoS ONE 2018, 13, e0206471. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Boetzer, M.; Pirovano, W. Toward almost closed genomes with GapFiller. Genome Biol. 2012, 13, R56. [Google Scholar] [CrossRef]
- Assefa, S.; Keane, T.M.; Otto, T.D.; Newbold, C.; Berriman, M. ABACAS: Algorithm-based automatic contiguation of assembled sequences. Bioinformatics 2009, 25, 1968–1969. [Google Scholar] [CrossRef]
- Tsai, I.J.; Otto, T.D.; Berriman, M. Improving draft assemblies by iterative mapping and assembly of short reads to eliminate gaps. Genome Biol. 2010, 11, R41. [Google Scholar] [CrossRef] [PubMed]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 27, 578–579. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Chevreux, B.; Pfisterer, T.; Drescher, B.; Driesel, A.J.; Müller, W.E.; Wetter, T.; Suhai, S. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004, 14, 1147–1159. [Google Scholar] [CrossRef]
- Altschup, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Carver, T.J.; Rutherford, K.M.; Berriman, M.; Rajandream, M.A.; Barrell, B.G.; Parkhill, J. ACT: The Artemis Comparison Tool. Bioinformatics 2005, 21, 3422–3423. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016, 44, D7–D19. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Bryant, S.H. CD-Search: Protein domain annotations on the fly. Nucleic Acids Res. 2004, 32, W327–W331. [Google Scholar] [CrossRef]
- Geer, L.Y.; Domrachev, M.; Lipman, D.J.; Bryant, S.H. CDART: Protein homology by domain architecture. Genome Res. 2002, 12, 1619–1623. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, K.; Parkhill, J.; Crook, J.; Horsnell, T.; Rice, P.; Rajandream, M.A.; Barrell, B. Artemis: Sequence visualization and annotation. Bioinformatics 2000, 16, 944–945. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Eichinger, V.; Nussbaumer, T.; Platzer, A.; Jehl, M.A.; Arnold, R.; Rattei, T. EffectiveDB--updates and novel features for a better annotation of bacterial secreted proteins and Type III, IV, VI secretion systems. Nucleic Acids Res. 2016, 44, D669–D674. [Google Scholar] [CrossRef]
- Treangen, T.J.; Ondov, B.D.; Koren, S.; Phillippy, A.M. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014, 15, 524. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- R Foundation for Statistical Computing. R Core Team R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019; Available online: https://www.r-project.org/ (accessed on 3 November 2021).
- Croucher, N.J.; Page, A.J.; Connor, T.R.; Delaney, A.J.; Keane, J.A.; Bentley, S.D.; Parkhill, J.; Harris, S.R. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2015, 43, e15. [Google Scholar] [CrossRef]
- Wilgenbusch, J.C.; Swofford, D. Inferring evolutionary trees with PAUP*. Curr. Protoc. Bioinf. 2003, 6, 6.4.1–6.4.28. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Croucher, N.J.; Goater, R.J.; Abudahab, K.; Aanensen, D.M.; Harris, S.R. Phandango: An interactive viewer for bacterial population genomics. Bioinformatics 2018, 34, 292–293. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Characteristics | Chlamydia pecorum Strain | ||||
|---|---|---|---|---|---|
| E58 | DBDeUG | DBDeUG_2018 | MC/MarsBar | MC/MarsBar_2018 | |
| Multilocus sequence typing a | ST23 | ST69 | ST69 | ||
| ompA genotype | E58 ompA | F | G | ||
| Collection year | Circa 1940 | 2010 | 2009 | ||
| Location | United States | Australia | Australia | ||
| Host | Cattle | Koala | Koala | ||
| Clinical presentation | Sporadic bovine encephalomyelitis | Urinary tract infection | Chronic cystitis | ||
| Anatomical site | Brain | Urogenital tract | Urogenital tract | ||
| BioSample | SAMN02603383 | SAMN02470821 | SAMN20256222 | SAMN02470823 | SAMN20256223 |
| GenBank accession numbers (chromosome and plasmid) | CP002608 | AZBB01000001 to AZBB01000008 | CP080401 & CP080402 | AZBC01000001 to AZBC01000014 | CP080403 & CP080404 |
| SRA accession | Not publicly available | Not publicly available | SRR15170908 | Not publicly available | SRR15170907 |
| Total No. of reads | Not applicable | 13,889,239 | 23,846,525 | 15,011,176 | 22,896,186 |
| No. of mapped reads | Not applicable | - | 16,458,765 | - | 13,336,552 |
| Chromosome length | 1,106,197 bp | 1,092,392 bp | 1,106,377 bp | 1,090,698 bp | 1,106,403 bp |
| Predicted CDS | 988 | 938 | 939 | 940 | 936 |
| GC content (%) | 41.11 | 41.12 | 41.12 | 41.12 | 41.11 |
| No. of tRNA genes | 38 | 38 | 38 | 38 | 38 |
| No. of rRNA operons | 3 | 3 | 3 | 3 | 3 |
| No. of pseudogenes | 1 | 1 | 2 | 2 | 3 |
| Plasmid length | Absent | 7547 bp | 7547 bp | 7547 bp | 7547 bp |
| Predicted CDS | Not applicable | 8 | 8 | 8 | 8 |
| GC content (%) | Not applicable | 31.55 | 31.55 | 31.55 | 31.55 |
| Tandem repeats | Not applicable | 4 × 22 bp | 4 × 22 bp | 4 × 22 bp | 4 × 22 bp |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
White, R.T.; Legione, A.R.; Taylor-Brown, A.; Fernandez, C.M.; Higgins, D.P.; Timms, P.; Jelocnik, M. Completing the Genome Sequence of Chlamydia pecorum Strains MC/MarsBar and DBDeUG: New Insights into This Enigmatic Koala (Phascolarctos cinereus) Pathogen. Pathogens 2021, 10, 1543. https://doi.org/10.3390/pathogens10121543
White RT, Legione AR, Taylor-Brown A, Fernandez CM, Higgins DP, Timms P, Jelocnik M. Completing the Genome Sequence of Chlamydia pecorum Strains MC/MarsBar and DBDeUG: New Insights into This Enigmatic Koala (Phascolarctos cinereus) Pathogen. Pathogens. 2021; 10(12):1543. https://doi.org/10.3390/pathogens10121543
Chicago/Turabian StyleWhite, Rhys T., Alistair R. Legione, Alyce Taylor-Brown, Cristina M. Fernandez, Damien P. Higgins, Peter Timms, and Martina Jelocnik. 2021. "Completing the Genome Sequence of Chlamydia pecorum Strains MC/MarsBar and DBDeUG: New Insights into This Enigmatic Koala (Phascolarctos cinereus) Pathogen" Pathogens 10, no. 12: 1543. https://doi.org/10.3390/pathogens10121543
APA StyleWhite, R. T., Legione, A. R., Taylor-Brown, A., Fernandez, C. M., Higgins, D. P., Timms, P., & Jelocnik, M. (2021). Completing the Genome Sequence of Chlamydia pecorum Strains MC/MarsBar and DBDeUG: New Insights into This Enigmatic Koala (Phascolarctos cinereus) Pathogen. Pathogens, 10(12), 1543. https://doi.org/10.3390/pathogens10121543