
Whole Genome Sequencing and Comparative Genome Analyses of Chlamydia abortus Strains of Avian Origin Suggests That Chlamydia abortus Species Should Be Expanded to Include Avian and Mammalian Subgroups

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Genomes of Avian C. abortus (Strains 15-70d24, 15-49d3 and 15-58d44, Representing Genotypes G1, G2, 1V, Respectively) Based on Hybrid Sequencing
2.2. Phenotypic Characterisation of Avian C. abortus Strains Corresponding to Genotypes G1, G2 and 1V (Strains 15-70d24, 15-49d3 and 15-58d44)
2.3. Genome Analysis
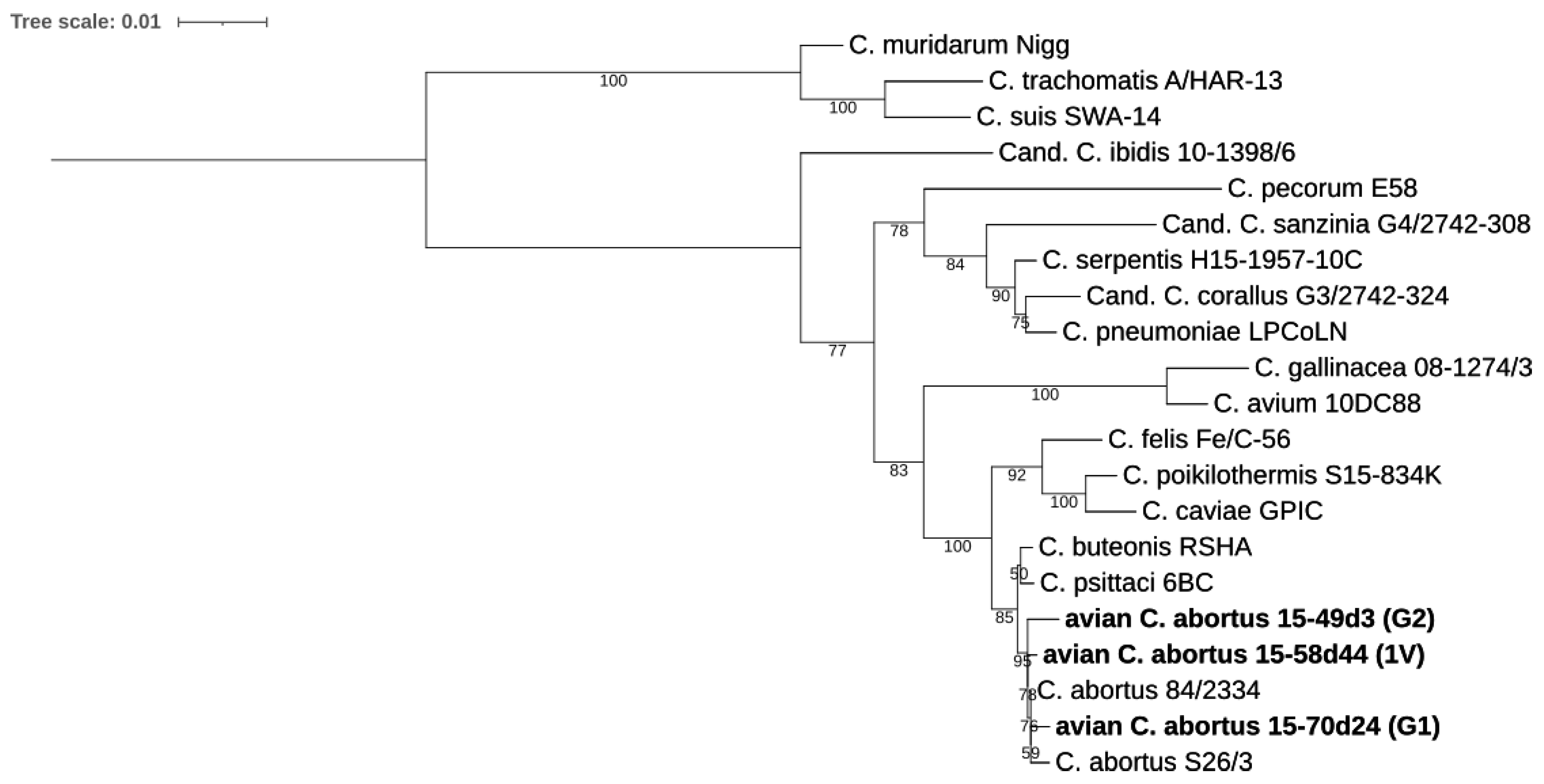
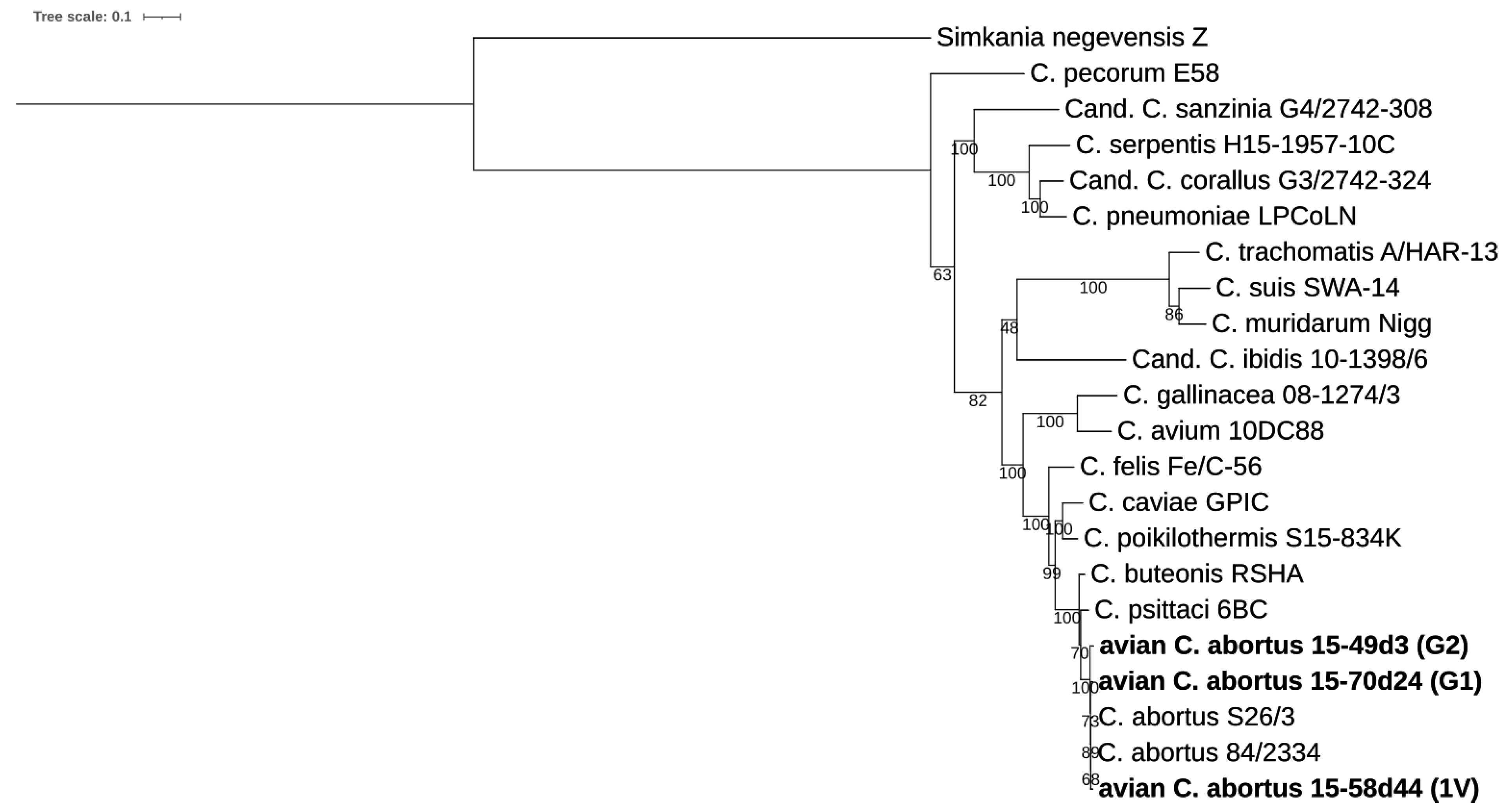
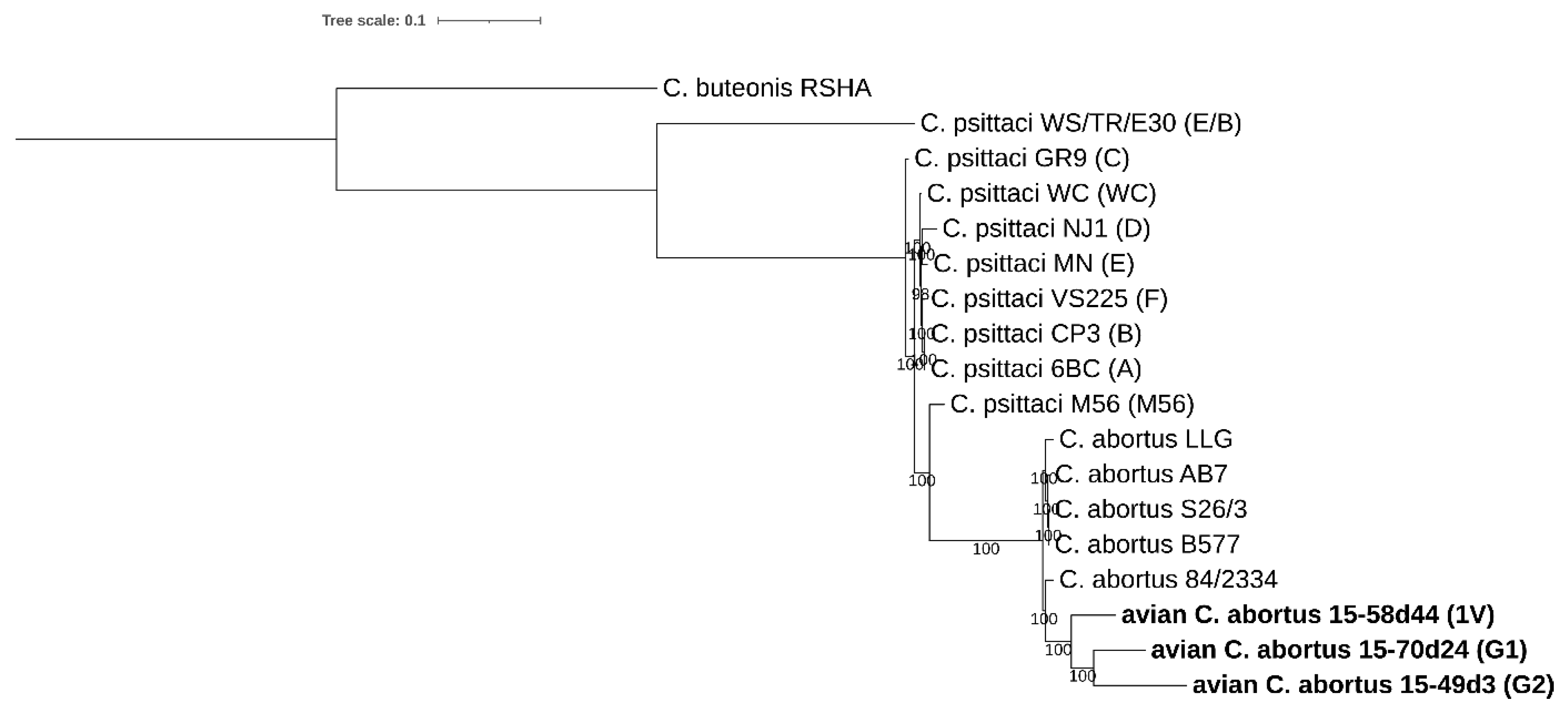
2.3.1. 16S rRNA and 23S rRNA Phylogenetic Analyses
2.3.2. Molecular Typing of Avian C. abortus Strains
2.3.3. ANIb and Tetra-Nucleotide Signatures
2.3.4. Plasmid Comparisons
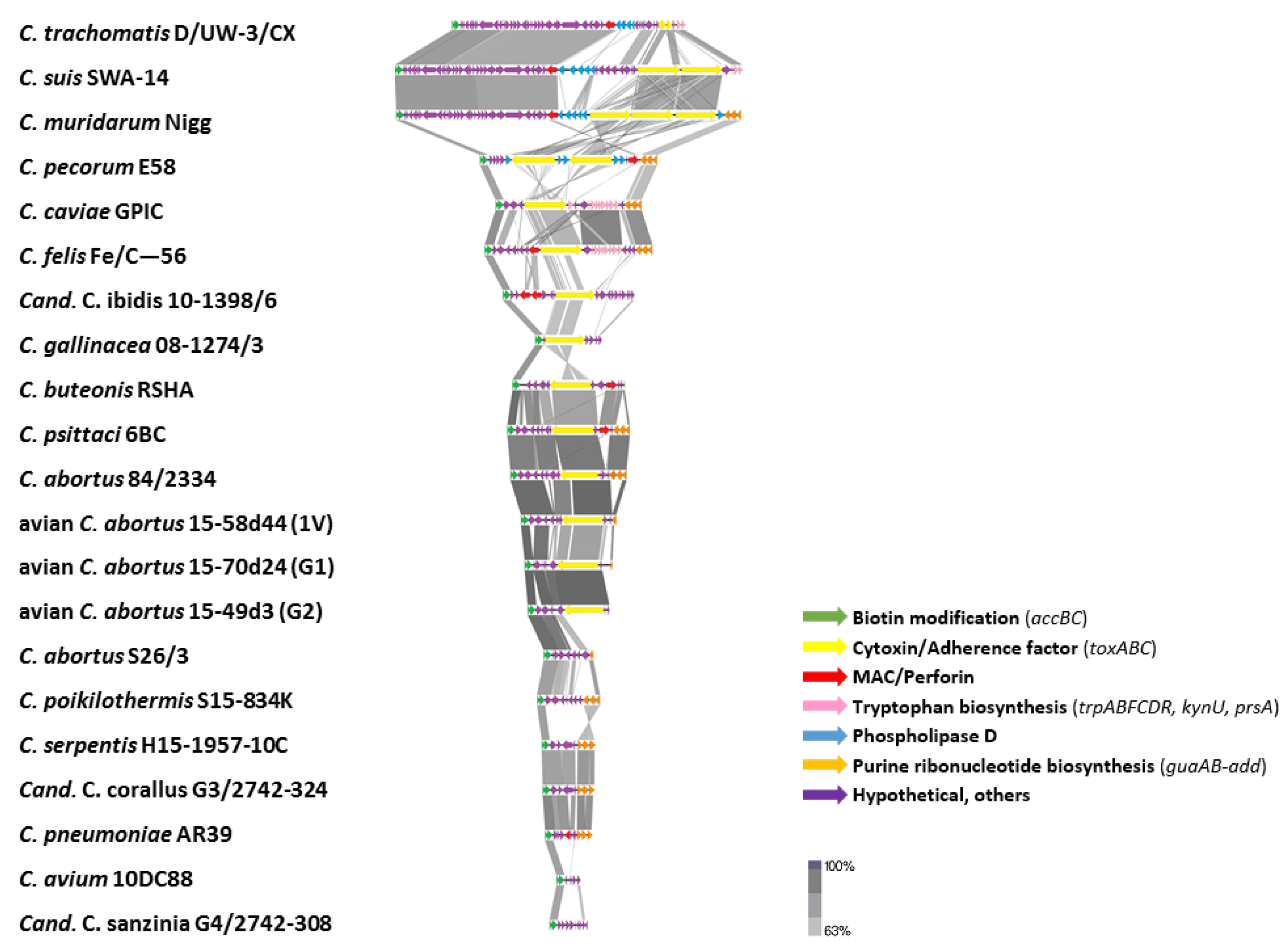
2.3.5. Plasticity Zone
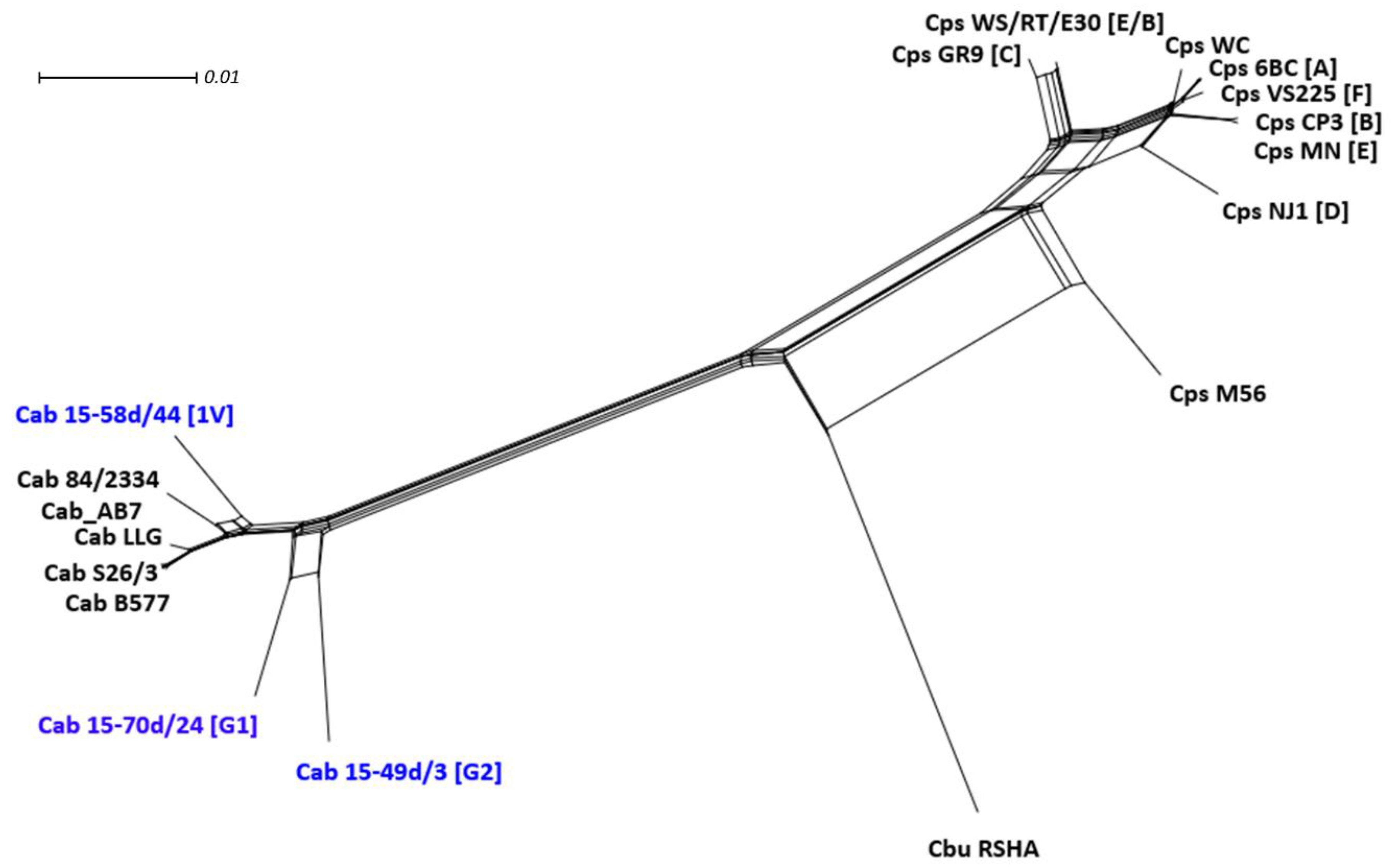
2.3.6. Phylogenetic Network Analysis
2.3.7. SNP Analysis
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains
4.2. Electron Microscopy
4.3. Genomic DNA Preparation
4.3.1. Illumina Sequencing
4.3.2. Nanopore (MinION) Sequencing
4.3.3. Quality Assessment and Quantification
4.4. Genome Sequencing, Assembly and Draft Annotation
Illumina and Nanopore Sequencing
4.5. Genome Analysis
4.5.1. 16S rRNA and 23S rRNA Phylogenetic Analyses
4.5.2. Molecular Typing of Avian C. abortus Strains
4.5.3. ANIb and Tetra-Nucleotide Signatures
4.5.4. Plasmid Comparisons
4.5.5. Plasticity Zone
4.5.6. Phylogenetic Network Analysis
4.5.7. SNP Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vorimore, F.; Hölzer, M.; Liebler-Tenorio, E.M.; Barf, L.M.; Delannoy, S.; Vittecoq, M.; Wedlarski, R.; Lécu, A.; Scharf, S.; Blanchard, Y.; et al. Evidence for the existence of a new genus Chlamydiifrater gen. nov. inside the family Chlamydiaceae with two new species isolated from flamingo (Phoenicopterus roseus): Chlamydiifrater phoenicopteri sp. nov. and Chlamydiifrater volucris sp. nov. Syst. Appl. Microbiol. 2021, 44, 126200. [Google Scholar] [CrossRef]
- Sachse, K.; Bavoil, P.M.; Kaltenboeck, B.; Stephens, R.S.; Kuo, C.C.; Rosselló-Móra, R.; Horn, M. Emendation of the family Chlamydiaceae: Proposal of a single genus, Chlamydia, to include all currently recognized species. Syst. Appl. Microbiol. 2015, 38, 99–103. [Google Scholar] [CrossRef]
- Staub, E.; Marti, H.; Biondi, R.; Levi, A.; Donati, M.; Leonard, C.A.; Ley, S.D.; Pillonel, T.; Greub, G.; Seth-Smith, H.M.B.; et al. Novel Chlamydia species isolated from snakes are temperature-sensitive and exhibit decreased susceptibility to azithromycin. Sci. Rep. 2018, 8, 5660. [Google Scholar] [CrossRef]
- Borel, N.; Greub, G. International committee on systematics of prokaryotes (ICSP) subcommittee on the taxonomy of chlamydiae. Minutes of the closed meeting, 5 July 2018 Woudschoten, Zeist, The Netherlands. Int. J. Syst. Evol. Microbiol. 2019, 69, 2606–2608. [Google Scholar] [CrossRef]
- Laroucau, K.; Vorimore, F.; Aaziz, R.; Solmonson, L.; Hsia, R.C.; Bavoil, P.M.; Fach, P.; Hölzer, M.; Wuenschmann, A.; Sachse, K. Chlamydia buteonis, a new Chlamydia species isolated from a red-shouldered hawk. Syst. Appl. Microbiol. 2019, 42, 125997. [Google Scholar] [CrossRef]
- Vorimore, F.; Hsia, R.C.; Huot-Creasy, H.; Bastian, S.; Deruyter, L.; Passet, A.; Sachse, K.; Bavoil, P.M.; Myers, G.; Laroucau, K. Isolation of a New Chlamydia species from the Feral Sacred Ibis Threskiornis aethiopicus Chlamydia ibidis. PLoS ONE 2013, 8, e74823. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Brown, A.; Bachmann, N.L.; Borel, N.; Polkinghorne, A. Culture-independent genomic characterisation of Candidatus Chlamydia sanzinia, a novel uncultivated bacterium infecting snakes. BMC Genom. 2016, 17, 10. [Google Scholar] [CrossRef]
- Taylor-Brown, A.; Spang, L.; Borel, N.; Polkinghorne, A. Culture-independent metagenomics supports discovery of uncultivable bacteria within the genus Chlamydia. Sci. Rep. 2017, 7, 10661. [Google Scholar] [CrossRef]
- Laroucau, K.; Ortega, N.; Vorimore, F.; Aaziz, R.; Mitura, A.; Szymańska-Czerwińska, M.; Cicerol, M.; Salinas, J.; Sachse, K.; Caro, M.R. Detection of a novel Chlamydia species in captive spur-thighed tortoises Testudo graeca in southeastern Spain and proposal of Candidatus Chlamydia testudinis. Syst. Appl. Microbiol. 2020, 43, 126071. [Google Scholar] [CrossRef] [PubMed]
- Borel, N.; Polkinghorne, A.; Pospischil, A. A Review on Chlamydial Diseases in Animals: Still a Challenge for Pathologists? Vet. Pathol. 2018, 55, 374–390. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, P.; Cao, X.; Lou, Z.; Zaręba-Marchewka, K.; Szymańska-Czerwińska, M.; Niemczuk, K.; Hu, B.; Bai, X.; Zhou, J. First report of Chlamydia abortus in farmed fur animals. Biomed Res. Int. 2018, 2018, 4289648. [Google Scholar] [CrossRef]
- Knittler, M.R.; Sachse, K. Chlamydia psittaci: Update on an underestimated zoonotic agent. Pathog. Dis. 2015, 73, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sachse, K.; Laroucau, K.; Vanrompay, D. Avian Chlamydiosis. Curr. Clin. Microbiol. Rep. 2015, 2, 10–21. [Google Scholar] [CrossRef]
- Szymańska-Czerwińska, M.; Niemczuk, K. Avian Chlamydiosis Zoonotic Disease. Vector-Borne Zoonotic Dis. 2016, 16, 1–3. [Google Scholar] [CrossRef]
- Pantchev, A.; Sting, R.; Bauerfeind, R.; Tyczka, J.; Sachse, K. New real-time PCR tests for species-specific detection of Chlamydophila psittaci and Chlamydophila abortus from tissue samples. Vet. J. 2009, 181, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Li, J.; Kaltenboeck, B.; Gong, J.; Fan, W.; Wang, C. Chlamydia gallinacea not C. psittaci is the endemic chlamydial species in chicken Gallus gallus. Sci. Rep. 2016, 6, 19638. [Google Scholar] [CrossRef]
- Szymańska-Czerwińska, M.; Mitura, A.; Zaręba, K.; Schnee, C.; Koncicki, A.; Niemczuk, K. Poultry in Poland as Chlamydiaceae carrier. J. Vet. Res. 2017, 61, 411–419. [Google Scholar] [CrossRef]
- Sachse, K.; Laroucau, K.; Riege, K.; Wehner, S.; Dilcher, M.; Huot, C.H.; Weidmann, M.; Myers, G.; Vorimore, F.; Vicari, N.; et al. Evidence for the existence of two new members of the family Chlamydiaceae and proposal of Chlamydia avium sp. nov. and Chlamydia gallinacea sp. nov. Syst. Appl. Microbiol. 2014, 37, 79–88. [Google Scholar] [CrossRef]
- Szymańska-Czerwińska, M.; Mitura, A.; Niemczuk, K.; Zaręba, K.; Jodełko, A.; Pluta, A.; Scharf, S.; Vitek, B.; Aaziz, R.; Vorimore, F.; et al. Dissemination and genetic diversity of chlamydial agents in Polish wildfowl: Isolation and molecular characterisation of avian Chlamydia abortus strains. PLoS ONE 2017, 12, e0174599. [Google Scholar] [CrossRef]
- Zaręba-Marchewka, K.; Szymańska-Czerwińska, M.; Niemczuk, K. Chlamydiae-What’s New? J. Vet. Res. 2020, 64, 461–467. [Google Scholar] [CrossRef]
- Longbottom, D.; Livingstone, M.; Ribeca, P.; Beeckman, D.S.A.; van der Ende, A.; Pannekoek, Y.; Vanrompay, D. Whole genome de novo sequencing and comparative genomic analyses suggests that Chlamydia psittaci strain 84/2334 should be reclassified as Chlamydia abortus species. BMC Genom. 2021, 22, 159. [Google Scholar] [CrossRef]
- Zaręba-Marchewka, K.; Szymańska-Czerwińska, M.; Mitura, A.; Niemczuk, K. Draft genome sequence of avian Chlamydia abortus genotype G1 strain 15-70d24, isolated from Eurasian teal in Poland. Microbiol. Resour. Announc. 2019, 8, e00658-19. [Google Scholar] [CrossRef]
- Zaręba-Marchewka, K.; Szymańska-Czerwińska, M.; Niemczuk, K. Draft Genome Sequences of Avian Chlamydia abortus Genotype G2 Strain 15-49d3, Isolated from Mallard, and Genotype 1V Strain 15-58d44, Isolated from Magpie in Poland. Microbiol. Resour. Announc. 2021, 4, e01203-19. [Google Scholar] [CrossRef]
- Pillonel, T.; Bertelli, C.; Salamin, N.; Greub, G. Taxogenomics of the order Chlamydiales. Int. J. Syst. Evol. Microbiol. 2015, 65, 1381–1393. [Google Scholar] [CrossRef]
- Holzer, M.; Laroucau, K.; Creasy, H.H.; Ott, S.; Vorimore, F.; Bavoil, P.M.; Sachse., K. Whole-genome sequence of Chlamydia gallinacea type strain 08-1274/3. Genome Announc. 2016, 4, e00708-16. [Google Scholar] [CrossRef] [PubMed]
- Rang, F.J.; Kloosterman, W.P.; De Ridder, J. From squiggle to basepair: Computational approaches for improving nanopore sequencing read accuracy. Genome Biol. 2018, 19, 90. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, S.; Beka, L.; Graf, J.; Klassen, J.L. Evaluation of strategies for the assembly of diverse bacterial genomes using MinION long-read sequencing. BMC Genom. 2019, 20, 23. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, N.L.; Polkinghorne, A.; Timms, P. Chlamydia genomics: Providing novel insights into chlamydial biology. Trends Microbiol. 2014, 22, 464–472. [Google Scholar] [CrossRef]
- Nunes, A.; Gomes, J.P. Evolution, phylogeny, and molecular epidemiology of Chlamydia. Infect. Genet. Evol. 2014, 23, 49–64. [Google Scholar] [CrossRef]
- Read, T.D.; Brunham, R.C.; Shen, C.; Gill, S.R.; Heidelberg, J.F.; White, O.; Hickey, E.K.; Peterson, J.; Utterback, T.; Berry, K.; et al. Genome sequences of Chlamydia trachomatis MoPn and Chlamydia pneumoniae AR39. Nucleic Acids Res. 2000, 28, 1397–1406. [Google Scholar] [CrossRef]
- Thomson, N.R.; Yeats, C.; Bell, K.; Holden, M.T.; Bentley, S.D.; Livingstone, M.; Cerdeño-Tárraga, A.M.; Harris, B.; Doggett, J.; Ormond, D.; et al. The Chlamydophila abortus genome sequence reveals an array of variable proteins that contribute to interspecies variation. Genome Res. 2005, 15, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Read, T.D.; Myers, G.S.; Brunham, R.C.; Nelson, W.C.; Paulsen, I.T.; Heidelberg, J.; Holtzapple, E.; Khouri, H.; Federova, N.B.; Carty, H.A.; et al. Genome sequence of Chlamydophila caviae (Chlamydia psittaci GPIC): Examining the role of niche-specific genes in the evolution of the Chlamydiaceae. Nucleic Acids Res. 2003, 31, 2134–2147. [Google Scholar] [CrossRef] [PubMed]
- Van Lent, S.; Piet, J.R.; Beeckman, D.; van der Ende, A.; Van Nieuwerburgh, F.; Bavoil, P.; Myers, G.; Vanrompay, D.; Pannekoek, Y. Full genome sequences of all nine Chlamydia psittaci genotype reference strains. J. Bacteriol. 2012, 194, 6930–6931. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, C.; Gong, S.; Hou, S.; Qi, M.; Liu, Q.; Baseman, J.; Zhong, G. Transformation of Chlamydia muridarum reveals a role for Pgp5 in suppression of plasmid-dependent gene expression. J. Bacteriol. 2014, 196, 989–998. [Google Scholar] [CrossRef]
- Jelocnik, M.; Bachmann, N.L.; Kaltenboeck, B.; Waugh, C.; Woolford, L.; Speight, K.N.; Gillett, A.; Higgins, D.P.; Flanagan, C.; Myers, G.S.; et al. Genetic diversity in the plasticity zone and the presence of the chlamydial plasmid differentiates Chlamydia pecorum strains from pigs, sheep, cattle, and koalas. BMC Genom. 2015, 16, 893. [Google Scholar] [CrossRef]
- Thomas, N.S.; Lusher, M.; Storey, C.C.; Clarke, I.N. Plasmid diversity in Chlamydia. Microbiol.-Sgm 1997, 143, 1847–1854. [Google Scholar] [CrossRef] [PubMed]
- Grinblat-Huse, V.; Drabek, E.F.; Creasy, H.H.; Daugherty, S.C.; Jones, K.M.; Santana-Cruz, I.; Tallon, L.J.; Read, T.D.; Hatch, T.P.; Bavoil, P.; et al. Genome sequences of the zoonotic pathogens Chlamydia psittaci 6BC and Cal10. J. Bacteriol. 2011, 193, 4039–4040. [Google Scholar] [CrossRef]
- Seth-Smith, H.M.; Harris, S.R.; Rance, R.; West, A.P.; Severin, J.A.; Ossewaarde, J.M.; Cutcliffe, L.T.; Skilton, R.J.; Marsh, P.; Parkhill, J.; et al. Genome sequence of the zoonotic pathogen Chlamydophila psittaci. J. Bacteriol. 2011, 193, 1282–1283. [Google Scholar] [CrossRef]
- Donati, M.; Huot-Creasy, H.; Humphrys, M.; Di Paolo, M.; Di Francesco, A.; Myers, G.S. Genome Sequence of Chlamydia suis MD56, Isolated from the Conjunctiva of a Weaned Piglet. Genome Announc. 2014, 2, e00425-14. [Google Scholar] [CrossRef] [PubMed]
- Hugall, A.; Timms, P.; Girjes, A.A.; Lavin, M.F. Conserved DNA sequences in Chlamydial plasmids. Plasmid 1989, 22, 91–98. [Google Scholar] [CrossRef]
- Pawlikowska-Warych, M.; Śliwa-Dominiak, J.; Deptuła, W. Chlamydial plasmids and bacteriophages. Acta Biochim. Pol. 2015, 62, 1–6. [Google Scholar] [CrossRef]
- Song, L.; Carlson, J.H.; Whitmire, W.M.; Kari, L.; Virtaneva, K.; Sturdevant, D.E.; Watkins, H.; Zhou, B.; Sturdevant, G.L.; Porcella, S.F.; et al. Chlamydia trachomatis Plasmid-Encoded Pgp4 Is a Transcriptional Regulator of Virulence-Associated Genes. Infect. Immun. 2013, 81, 636–644. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.J.; Li, B.; Ghonasgi, T.; Haase, C.P.; Qin, Z.S.; Dean, D.; Read, T.D. Direct amplification, sequencing and profiling of Chlamydia trachomatis strains in single and mixed infection clinical samples. PLoS ONE 2014, 9, e99290. [Google Scholar] [CrossRef] [PubMed]
- Szabo, K.V.; O’Neill, C.E.; Clarke, I.N. Diversity in Chlamydial plasmids. PLoS ONE 2020, 15, e0233298. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.; Quigley, B.L.; Timms, P. Seventy Years of Chlamydia Vaccine Research-Limitations of the Past and Directions for the Future. Front. Microbiol. 2019, 10, 70. [Google Scholar] [CrossRef] [PubMed]
- Greub, G.; Bavoil, P. International Committee on Systematics of Prokaryotes Subcommittee on the taxonomy of Chlamydiae. Minutes of the closed meeting, 7 September 2016, Oxford, UK. Int. J. Syst. Evol. Microbiol. 2018, 68, 3683–3684. [Google Scholar] [CrossRef]
- Van Loock, M.; Vanrompay, D.; Herrmann, B.; Vander Stappen, J.; Volckaert, G.; Goddeeris, B.M.; Everett, K.D.E. Missing links in the divergence of Chlamydophila abortus from Chlamydophila psittaci. Int. J. Syst. Evol. Microbiol. 2003, 53, 761–770. [Google Scholar] [CrossRef]
- Pannekoek, Y.; Dickx, V.; Beeckman, D.S.; Jolley, K.A.; Keijzers, W.C.; Vretou, E.; Maiden, M.C.; Vanrompay, D.; van der Ende, A. Multi locus sequence typing of Chlamydia reveals an association between Chlamydia psittaci genotypes and host species. PLoS ONE 2010, 5, e14179. [Google Scholar] [CrossRef]
- Ehricht, R.; Slickers, P.; Goellner, S.; Hotzel, H.; Sachse, K. Optimized DNA microarray assay allows detection and genotyping of single PCR-amplifiable target copies. Mol. Cell. Probe. 2006, 20, 60–63. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Bushnell, B. BBTools Software Package. 2014. Available online: http://bbtools.jgi.doe.gov (accessed on 17 August 2021).
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Cuccuru, G.; Orsini, M.; Pinna, A.; Sbardellati, A.; Soranzo, N.; Travaglione, A.; Uva, P.; Zanetti, G.; Fotia, G. Orione, a web-based framework for NGS analysis in microbiology. Bioinformatics 2014, 30, 1928–1929. [Google Scholar] [CrossRef] [PubMed]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, 537–544. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 8, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection foraccurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Garrison, E.; Kronenberg, Z.N.; Dawson, E.T.; Pedersen, B.S.; Prins, P. Vcflib and tools for processing the VCF variant call format. BioRxiv 2021. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, 12. [Google Scholar] [CrossRef]
- Danecek, P.; McCarthy, S.A. BCFtools/csq: Haplotype-aware variant consequences. Bioinformatics 2017, 33, 2037–2039. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaręba-Marchewka, K.; Szymańska-Czerwińska, M.; Livingstone, M.; Longbottom, D.; Niemczuk, K. Whole Genome Sequencing and Comparative Genome Analyses of Chlamydia abortus Strains of Avian Origin Suggests That Chlamydia abortus Species Should Be Expanded to Include Avian and Mammalian Subgroups. Pathogens 2021, 10, 1405. https://doi.org/10.3390/pathogens10111405
Zaręba-Marchewka K, Szymańska-Czerwińska M, Livingstone M, Longbottom D, Niemczuk K. Whole Genome Sequencing and Comparative Genome Analyses of Chlamydia abortus Strains of Avian Origin Suggests That Chlamydia abortus Species Should Be Expanded to Include Avian and Mammalian Subgroups. Pathogens. 2021; 10(11):1405. https://doi.org/10.3390/pathogens10111405
Chicago/Turabian StyleZaręba-Marchewka, Kinga, Monika Szymańska-Czerwińska, Morag Livingstone, David Longbottom, and Krzysztof Niemczuk. 2021. "Whole Genome Sequencing and Comparative Genome Analyses of Chlamydia abortus Strains of Avian Origin Suggests That Chlamydia abortus Species Should Be Expanded to Include Avian and Mammalian Subgroups" Pathogens 10, no. 11: 1405. https://doi.org/10.3390/pathogens10111405
APA StyleZaręba-Marchewka, K., Szymańska-Czerwińska, M., Livingstone, M., Longbottom, D., & Niemczuk, K. (2021). Whole Genome Sequencing and Comparative Genome Analyses of Chlamydia abortus Strains of Avian Origin Suggests That Chlamydia abortus Species Should Be Expanded to Include Avian and Mammalian Subgroups. Pathogens, 10(11), 1405. https://doi.org/10.3390/pathogens10111405