The Nucleation and the Intrinsic Microstructure Evolution of Martensite from
332
〈
113
〉
β
Twin Boundary in β Titanium: First-Principles Calculations



Abstract
1. Introduction
2. Computational Methods and Model
2.1. Computational Methods
2.2. Twin Boundary Model
3. Results and Discussions
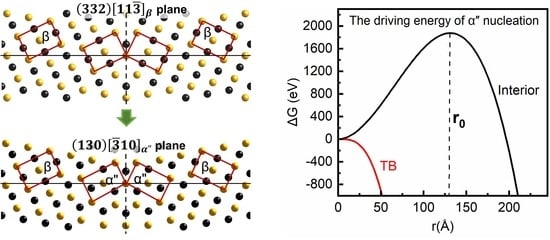
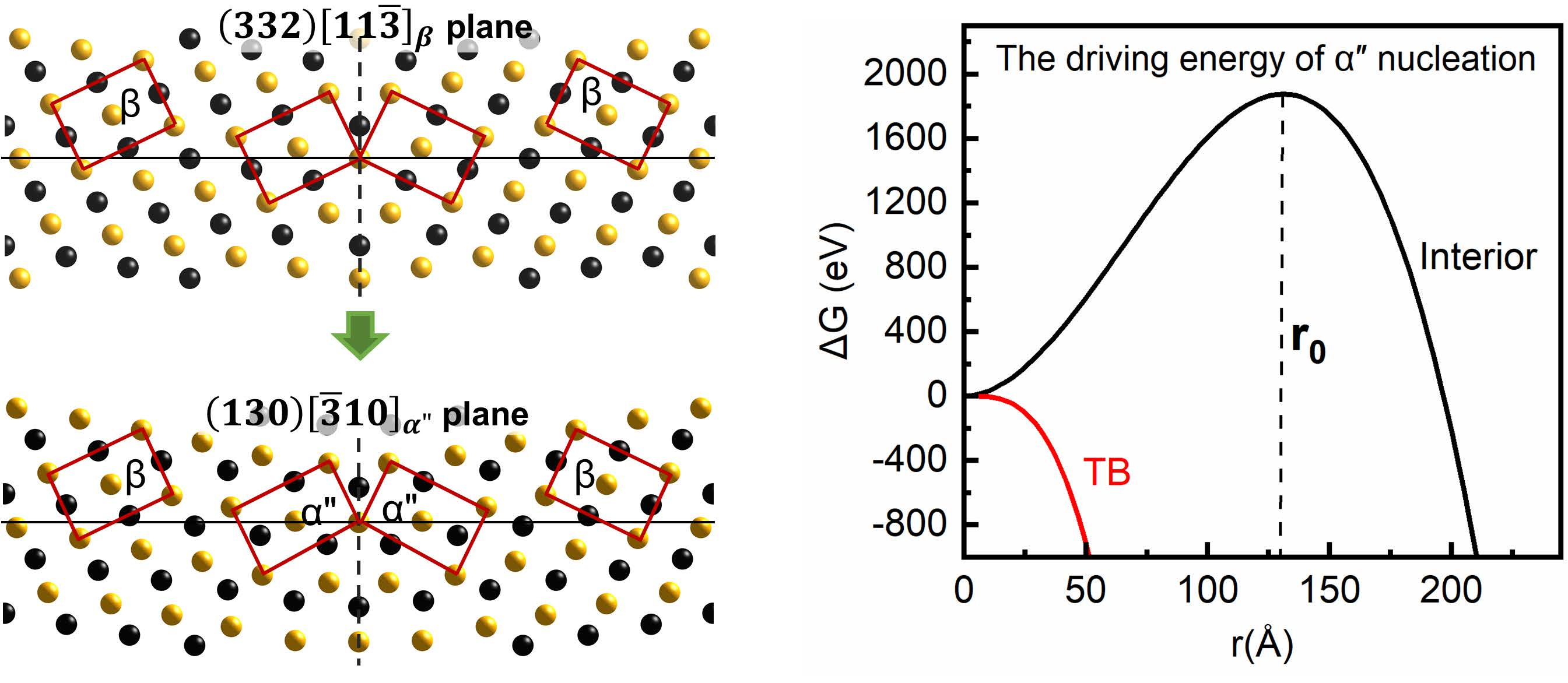
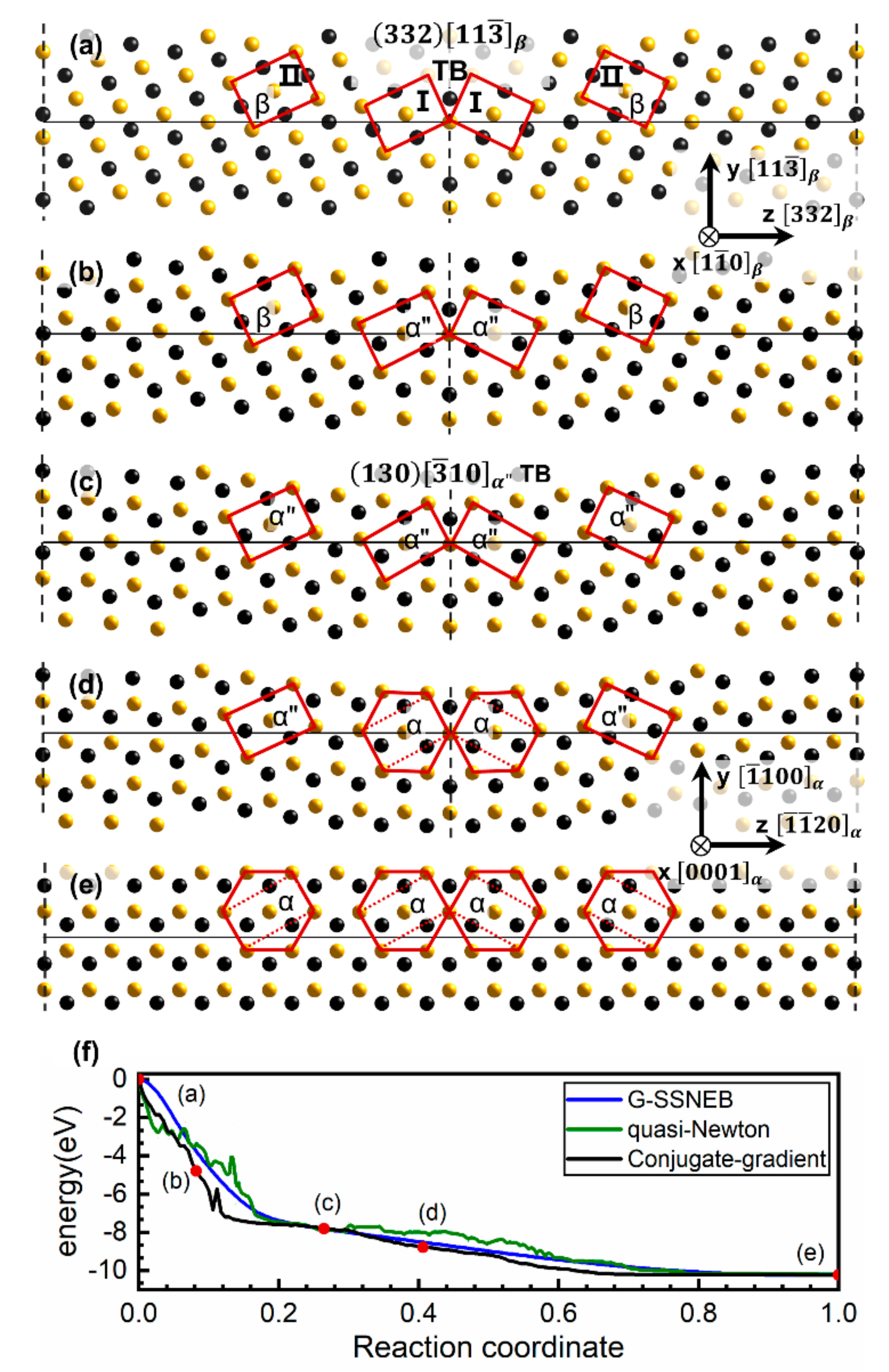
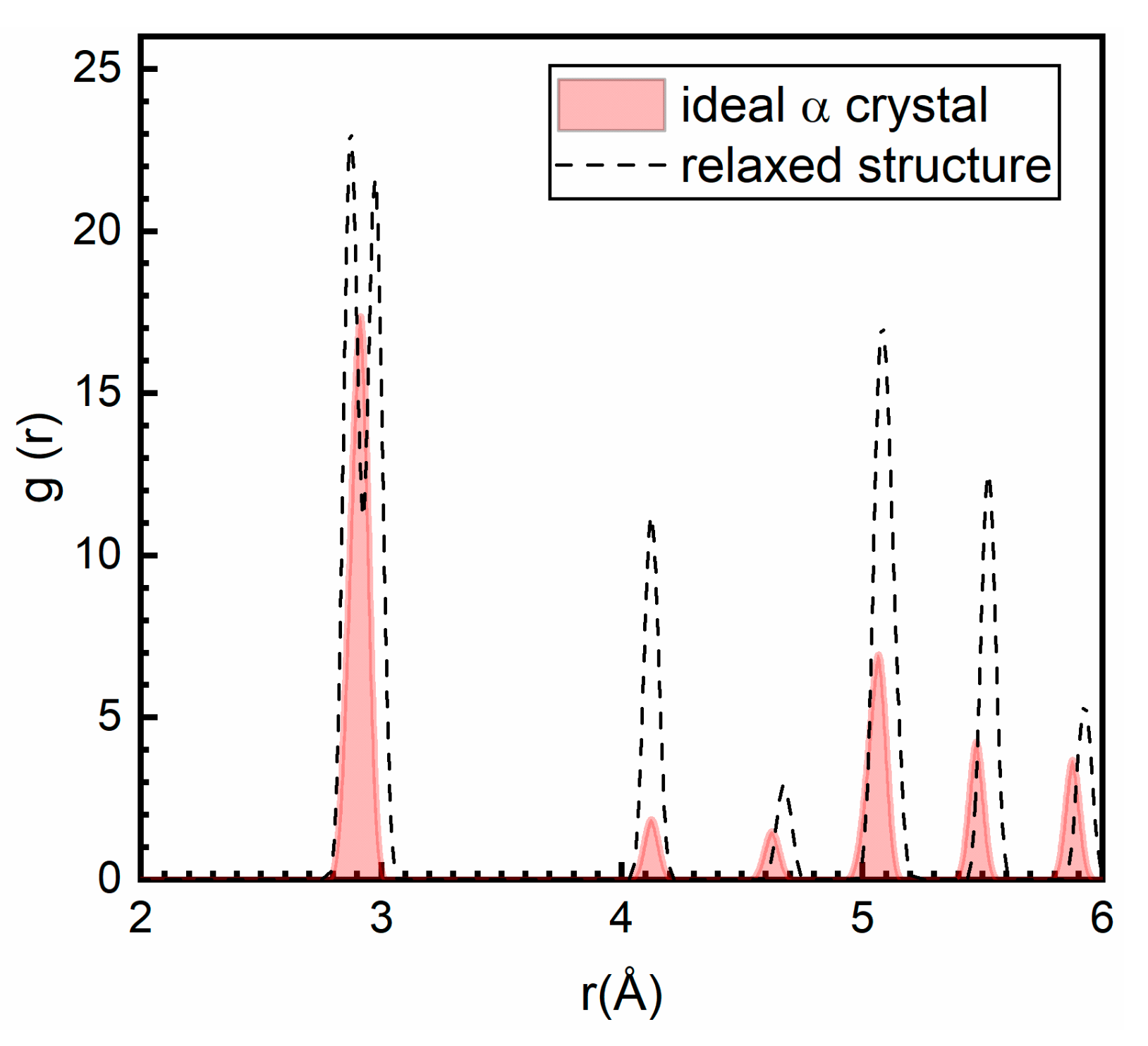
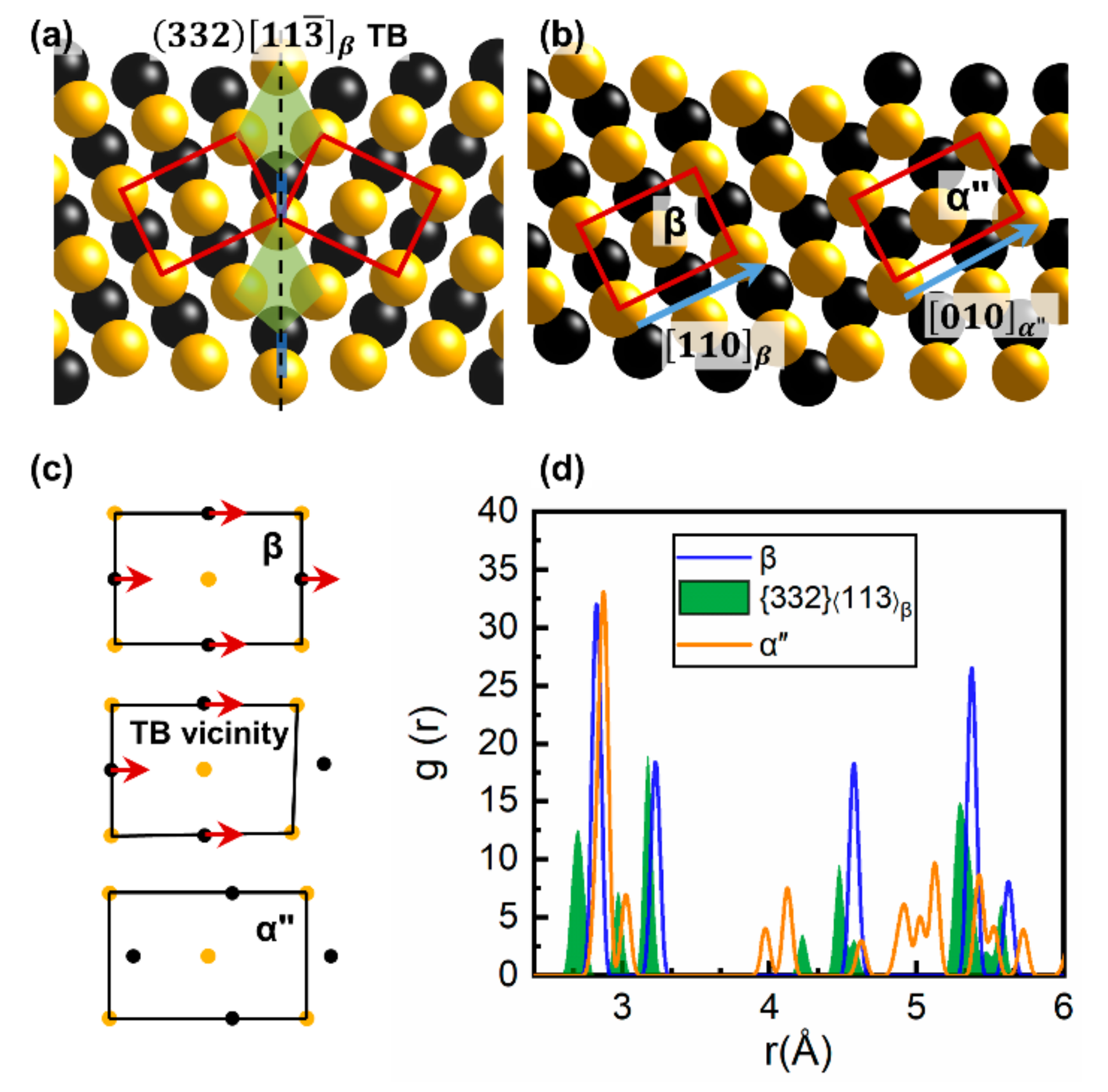
3.1. The Intrinsic Evolution from TB to α Phase
3.2. The Different Displacements of Atoms Close to and Far Away from TB
3.3. The Nucleation and Structure Evolution of Martensite from Crystallographic Perspective
3.4. The Nucleation and Structure Evolution of Martensite from Kinetic Calculations
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Niinomi, M. Recent research and development in titanium alloys for biomedical applications and healthcare goods. Sci. Technol. Adv. Mater. 2003, 4, 445–454. [Google Scholar] [CrossRef]
- Banerjee, D.; Williams, J.C. Perspectives on Titanium Science and Technology. Acta Mater. 2013, 61, 844–879. [Google Scholar] [CrossRef]
- Weiss, I.; Semiatin, S.L. Thermomechanical processing of beta titanium alloys—An overview. Mater. Sci. Eng. A 1998, 243, 46–65. [Google Scholar] [CrossRef]
- Williams, J.C.; Starke, E.A. Progress in structural materials for aerospace systems11The Golden Jubilee Issue—Selected topics in Materials Science and Engineering: Past, Present and Future, edited by S. Suresh. Acta Mater. 2003, 51, 5775–5799. [Google Scholar] [CrossRef]
- Grosdidier, T.; Philippe, M.J. Deformation induced martensite and superelasticity in a β-metastable titanium alloy. Mater. Sci. Eng. A 2000, 291, 218–223. [Google Scholar] [CrossRef]
- Min, X.H.; Tsuzaki, K.; Emura, S.; Tsuchiya, K. Enhancement of uniform elongation in high strength Ti–Mo based alloys by combination of deformation modes. Mater. Sci. Eng. A 2011, 528, 4569–4578. [Google Scholar] [CrossRef]
- Marteleur, M.; Sun, F.; Gloriant, T.; Vermaut, P.; Jacques, P.J.; Prima, F. On the design of new β-metastable titanium alloys with improved work hardening rate thanks to simultaneous TRIP and TWIP effects. Scr. Mater. 2012, 66, 749–752. [Google Scholar] [CrossRef]
- Ahmed, M.; Wexler, D.; Casillas, G.; Savvakin, D.G.; Pereloma, E.V. Strain rate dependence of deformation-induced transformation and twinning in a metastable titanium alloy. Acta Mater. 2016, 104, 190–200. [Google Scholar] [CrossRef]
- Gao, J.; Huang, Y.; Guan, D.; Knowles, A.J.; Ma, L.; Dye, D.; Rainforth, W.M. Deformation mechanisms in a metastable beta titanium twinning induced plasticity alloy with high yield strength and high strain hardening rate. Acta Mater. 2018, 152, 301–314. [Google Scholar] [CrossRef]
- Sun, F.; Zhang, J.Y.; Marteleur, M.; Brozek, C.; Rauch, E.F.; Veron, M.; Vermaut, P.; Jacques, P.J.; Prima, F. A new titanium alloy with a combination of high strength, high strain hardening and improved ductility. Scripta Mater. 2015, 94, 17–20. [Google Scholar] [CrossRef]
- Ahmed, M.; Gazder, A.A.; Saleh, A.A.; Wexler, D.; Pereloma, E.V. Stress-Induced Twinning and Phase Transformations during the Compression of a Ti-10V-3Fe-3Al Alloy. Metall. Mater. Trans. A 2017, 48, 2791–2800. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Li, J.S.; Chen, Z.; Meng, Q.K.; Sun, F.; Shen, B.L. Microstructural evolution of a ductile metastable β titanium alloy with combined TRIP/TWIP effects. J. Alloys Compd. 2017, 699, 775–782. [Google Scholar] [CrossRef]
- Sun, F.; Zhang, J.Y.; Marteleur, M.; Gloriant, T.; Vermaut, P.; Laillé, D.; Castany, P.; Curfs, C.; Jacques, P.J.; Prima, F. Investigation of early stage deformation mechanisms in a metastable β titanium alloy showing combined twinning-induced plasticity and transformation-induced plasticity effects. Acta Mater. 2013, 61, 6406–6417. [Google Scholar] [CrossRef]
- Castany, P.; Yang, Y.; Bertrand, E.; Gloriant, T. Reversion of a Parent {130} 〈310〉 α″ Martensitic Twinning System at the Origin of {332} 〈113〉 β Twins Observed in Metastable β Titanium Alloys. Phys. Rev. Lett. 2016, 117, 245501. [Google Scholar] [CrossRef]
- Lilensten, L.; Danard, Y.; Brozek, C.; Mantri, S.; Castany, P.; Gloriant, T.; Vermaut, P.; Sun, F.; Banerjee, R.; Prima, F. On the heterogeneous nature of deformation in a strain-transformable beta metastable Ti-V-Cr-Al alloy. Acta Mater. 2019, 162, 268–276. [Google Scholar] [CrossRef]
- Ahmed, M.; Wexler, D.; Casillas, G.; Ivasishin, O.M.; Pereloma, E.V. The influence of beta phase stability on deformation mode and compressive mechanical properties of Ti-10V-3Fe-3Al alloy. Acta Mater. 2015, 84, 124–135. [Google Scholar] [CrossRef]
- Chen, B.; Sun, W. Transitional structure of {332}β twin boundary in a deformed metastable β-type Ti-Nb-based alloy, revealed by atomic resolution electron microscopy. Scr. Mater. 2018, 150, 115–119. [Google Scholar] [CrossRef]
- Lai, M.J.; Tasan, C.C.; Raabe, D. On the mechanism of {332} twinning in metastable β titanium alloys. Acta Mater. 2016, 111, 173–186. [Google Scholar] [CrossRef]
- Raabe, D.; Sander, B.; Friak, M.; Ma, D.; Neugebauer, J. Theory-guided bottom-up design of beta-titanium alloys as biomaterials based on first principles calculations: Theory and experiments. Acta Mater. 2007, 55, 4475–4487. [Google Scholar] [CrossRef]
- Zhou, W.C.; Sahara, R.; Tsuchiya, K. First-principles study of the phase stability and elastic properties of Ti-X alloys (X = Mo, Nb, Al, Sn, Zr, Fe, Co, and O). J. Alloys Compd. 2017, 727, 579–595. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal--amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Sheppard, D.; Xiao, P.; Chemelewski, W.; Johnson, D.D.; Henkelman, G. A generalized solid-state nudged elastic band method. J. Chem. Phys. 2012, 136, 074103. [Google Scholar] [CrossRef]
- Han, J.; Vitek, V.; Srolovitz, D.J. The grain-boundary structural unit model redux. Acta Mater. 2017, 133, 186–199. [Google Scholar] [CrossRef]
- Sutton, A.P.; Vitek, V. On the structure of tilt grain boundaries in cubic metals I. Symmetrical tilt boundaries. Philos. Trans. R. Soc. A-Math. Phys. Eng. 1983, 309, 1–36. [Google Scholar] [CrossRef]
- Tschopp, M.A.; Solanki, K.N.; Gao, F.; Sun, X.; Khaleel, M.A.; Horstemeyer, M.F. Probing grain boundary sink strength at the nanoscale: Energetics and length scales of vacancy and interstitial absorption by grain boundaries in alpha-Fe. Phys. Rev. B 2012, 85, 064108. [Google Scholar] [CrossRef]
- Bhattacharya, S.K.; Tanaka, S.; Shiihara, Y.; Kohyama, M. Ab initio study of symmetrical tilt grain boundaries in bcc Fe: Structural units, magnetic moments, interfacial bonding, local energy and local stress. J. Phys. Condens. Matter 2013, 25, 135004. [Google Scholar] [CrossRef]
- Hahn, E.N.; Fensin, S.J.; Germann, T.C.; Meyers, M.A. Symmetric tilt boundaries in body-centered cubic tantalum. Scr. Mater. 2016, 116, 108–111. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Pack, J.D.; Monkhorst, H.J. “Special points for Brillouin-zone integrations”—A reply. Phys. Rev. B 1977, 16, 1748–1749. [Google Scholar] [CrossRef]
- Joost, W.J.; Ankem, S.; Kuklja, M.M. Interaction between oxygen interstitials and deformation twins in alpha-titanium. Acta Mater. 2016, 105, 44–51. [Google Scholar] [CrossRef]
- Kumar, A.; Wang, J.; Tome, C.N. First-principles study of energy and atomic solubility of twinning-associated boundaries in hexagonal metals. Acta Mater. 2015, 85, 144–154. [Google Scholar] [CrossRef]
- Mei, Z.-G.; Shang, S.-L.; Wang, Y.; Liu, Z.-K. Density-functional study of the thermodynamic properties and the pressure--temperature phase diagram of Ti. Phys. Rev. B 2009, 80, 104116. [Google Scholar] [CrossRef]
- Persson, K.; Ekman, M.; Ozolins, V. Phonon instabilities in bcc Sc, Ti, La, and Hf. Phys. Rev. B 2000, 61, 11221–11224. [Google Scholar] [CrossRef]
- Grimvall, G.; Magyari-Kope, B.; Ozolins, V.; Persson, K.A. Lattice instabilities in metallic elements. Rev. Mod. Phys. 2012, 84, 945–986. [Google Scholar] [CrossRef]
- Vohra, Y.K.; Spencer, P.T. Novel gamma-phase of titanium metal at megabar pressures. Phys. Rev. Lett. 2001, 86, 3068–3071. [Google Scholar] [CrossRef]
- Zimmermann, F.; Humbert, M. Determination of the habit plane characteristics in the β–α′ phase transformation induced by stress in Ti–5Al–2Sn–4Zr–4Mo–2Cr–1Fe. Acta Mater. 2002, 50, 1735–1740. [Google Scholar] [CrossRef]
- Li, C.-X.; Luo, H.-B.; Hu, Q.-M.; Yang, R.; Yin, F.-X.; Umezawa, O.; Vitos, L. Lattice parameters and relative stability of α″ phase in binary titanium alloys from first-principles calculations. Solid State Commun. 2013, 159, 70–75. [Google Scholar] [CrossRef]
- Kim, H.Y.; Ikehara, Y.; Kim, J.I.; Hosoda, H.; Miyazaki, S. Martensitic transformation, shape memory effect and superelasticity of Ti–Nb binary alloys. Acta Mater. 2006, 54, 2419–2429. [Google Scholar] [CrossRef]
- Bertrand, E.; Castany, P.; Yang, Y.; Menou, E.; Gloriant, T. Deformation twinning in the full-α″ martensitic Ti–25Ta–20Nb shape memory alloy. Acta Mater. 2016, 105, 94–103. [Google Scholar] [CrossRef]
- Mantri, S.A.; Sun, F.; Choudhuri, D.; Alam, T.; Gwalani, B.; Prima, F.; Banerjee, R. Deformation Induced Hierarchical Twinning Coupled with Omega Transformation in a Metastable β-Ti Alloy. Sci. Rep. 2019, 9, 1334. [Google Scholar] [CrossRef] [PubMed]
- Drossinos, Y.; Kevrekidis, P.G. Classical nucleation theory revisited. Phys. Rev. E 2003, 67, 026127. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wan, W.; Zhu, L.; Jiang, Y.; Shao, S.; Yang, G.; Liu, H.; Yi, D.; Cao, S.; Hu, Q. Experimental and DFT characterization of interphase boundaries in titanium and the implications for ω-assisted α phase precipitation. Acta Mater. 2018, 151, 406–415. [Google Scholar] [CrossRef]
- Yin, J.O.; Chen, G.; Zhao, S.Y.; Ge, Y.; Li, Z.F.; Yang, P.J.; Han, W.Z.; Wang, J.; Tang, H.P.; Cao, P. Microstructural characterization and properties of Ti-28Ta at.% powders produced by plasma rotating electrode process. J. Alloys Compd. 2017, 713, 222–228. [Google Scholar] [CrossRef]
- Frutos, E.; Karlík, M.; Jiménez, J.A.; Langhansová, H.; Lieskovská, J.; Polcar, T. Development of new β/α″-Ti-Nb-Zr biocompatible coating with low Young’s modulus and high toughness for medical applications. Mater. Des. 2018, 142, 44–55. [Google Scholar] [CrossRef]
- Yao, T.; Du, K.; Wang, H.; Huang, Z.; Li, C.; Li, L.; Hao, Y.; Yang, R.; Ye, H. In situ scanning and transmission electron microscopy investigation on plastic deformation in a metastable β titanium alloy. Acta Mater. 2017, 133, 21–29. [Google Scholar] [CrossRef]
- Furuhara, T.; Nakamori, H.; Maki, T. Crystallography of α Phase Precipitated on Dislocations and Deformation Twin Boundaries in a β Titanium Alloy. Mater. Trans. JIM 1992, 33, 585–595. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interfaces | Lattice Parameters (Å) | Interface Energy (mJ/m2) | ||
|---|---|---|---|---|
| a | b | c | ||
| 4.59 | 5.39 | 50.66 | 777.38 | |
| 4.59 | 10.62 | 37.45 | 76.71 | |
| 3.15 | 4.59 | 37.93 | 740.04 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Q.-J.; Ma, S.-Y.; Wang, S.-Q.
The Nucleation and the Intrinsic Microstructure Evolution of Martensite from
Chen Q-J, Ma S-Y, Wang S-Q.
The Nucleation and the Intrinsic Microstructure Evolution of Martensite from
Chen, Qiu-Jie, Shang-Yi Ma, and Shao-Qing Wang.
2019. "The Nucleation and the Intrinsic Microstructure Evolution of Martensite from
Chen, Q.-J., Ma, S.-Y., & Wang, S.-Q.
(2019). The Nucleation and the Intrinsic Microstructure Evolution of Martensite from