Large-Scale Molecular Dynamics Simulations of Homogeneous Nucleation of Pure Aluminium




Abstract
1. Introduction
2. Simulation Methodology
3. Results and Discussion
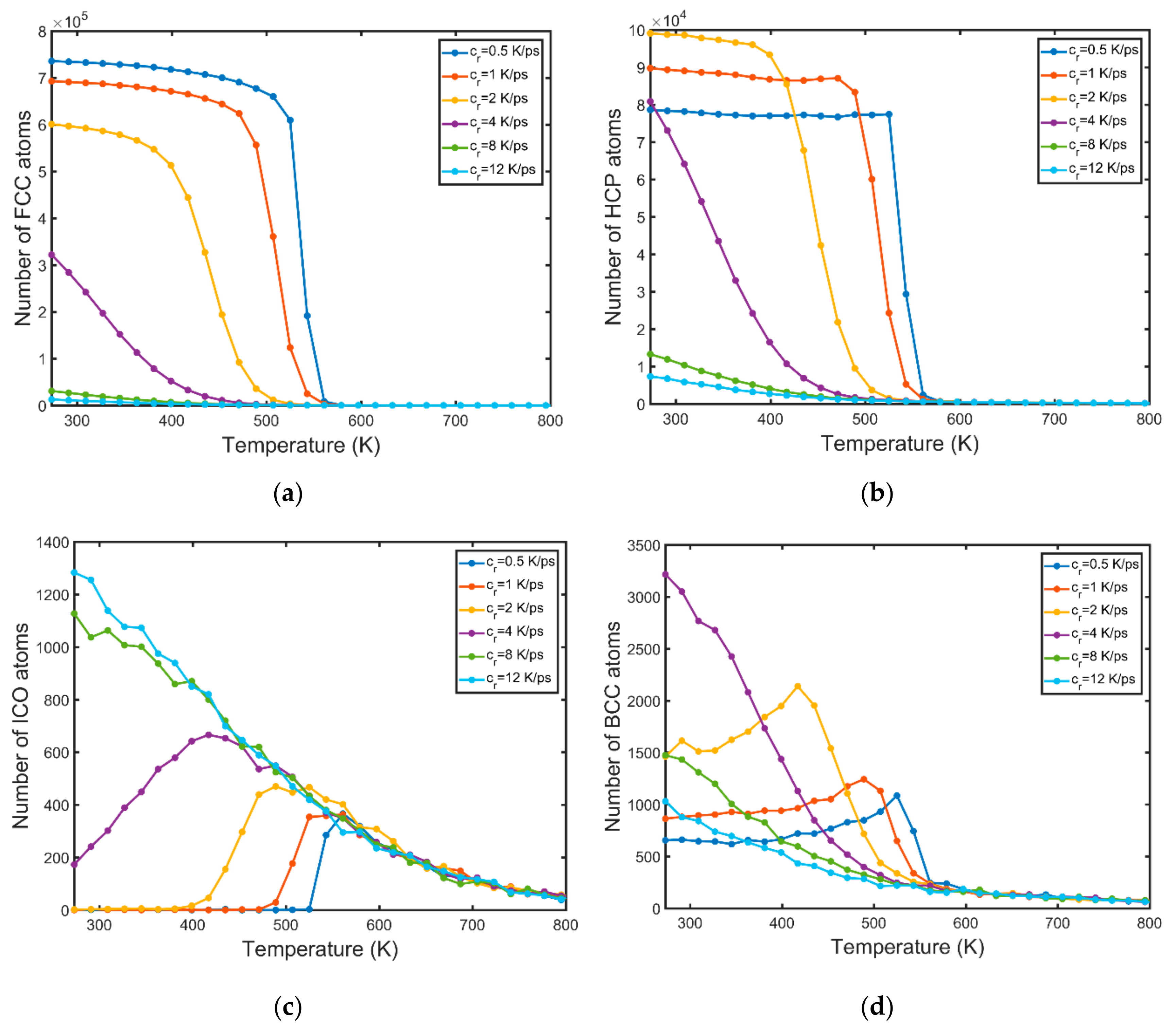
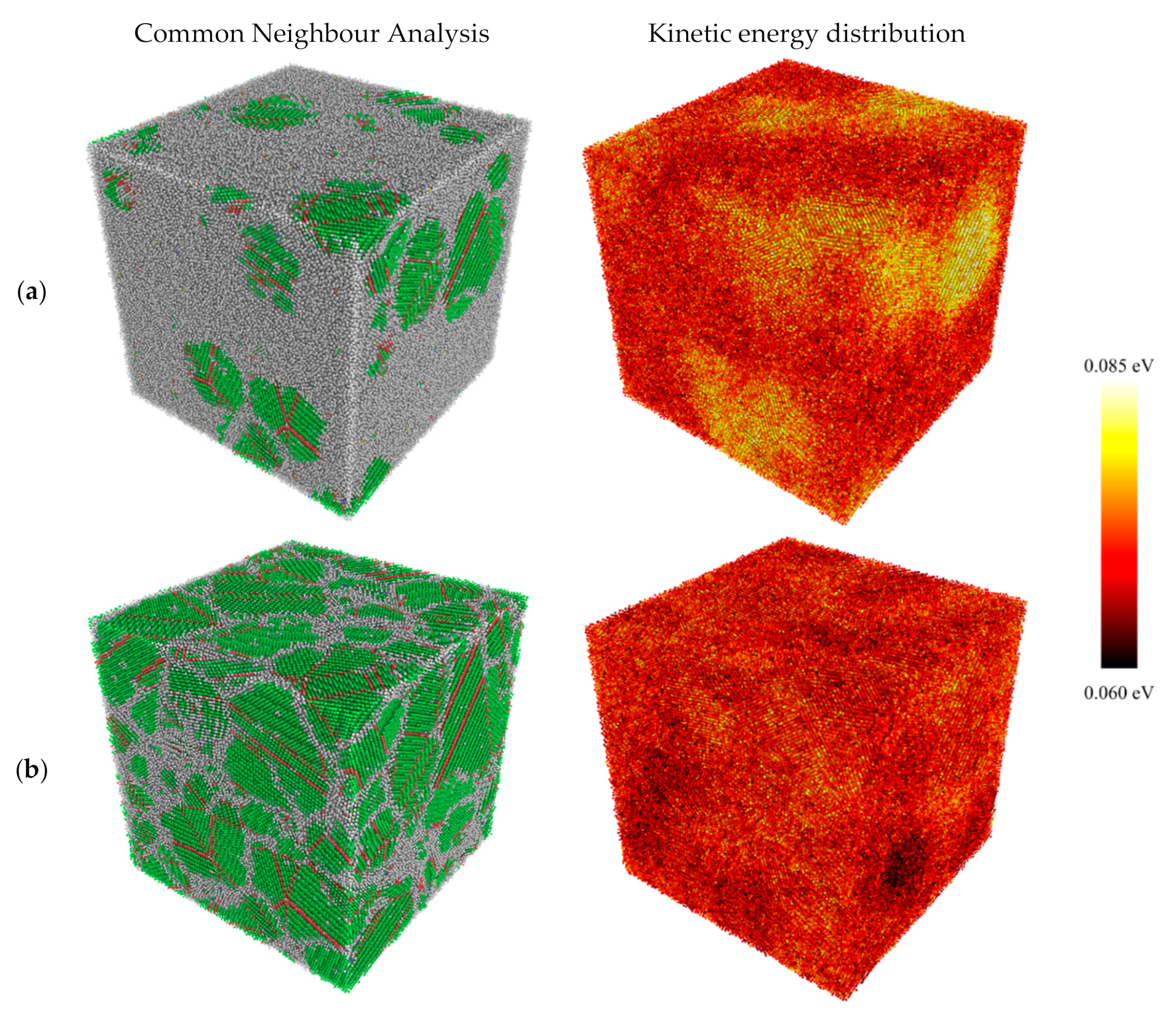
3.1. Structural Properties
3.2. Nucleation
3.3. Kinetic Energy and Temperature
4. Conclusions
- The BCC phase is formed prior to the FCC and HCP phases. A high number of BCC atoms is indicative of high nucleation and growth rates.
- Solidification occurs in a wider range of temperatures for higher values of the cooling rate.
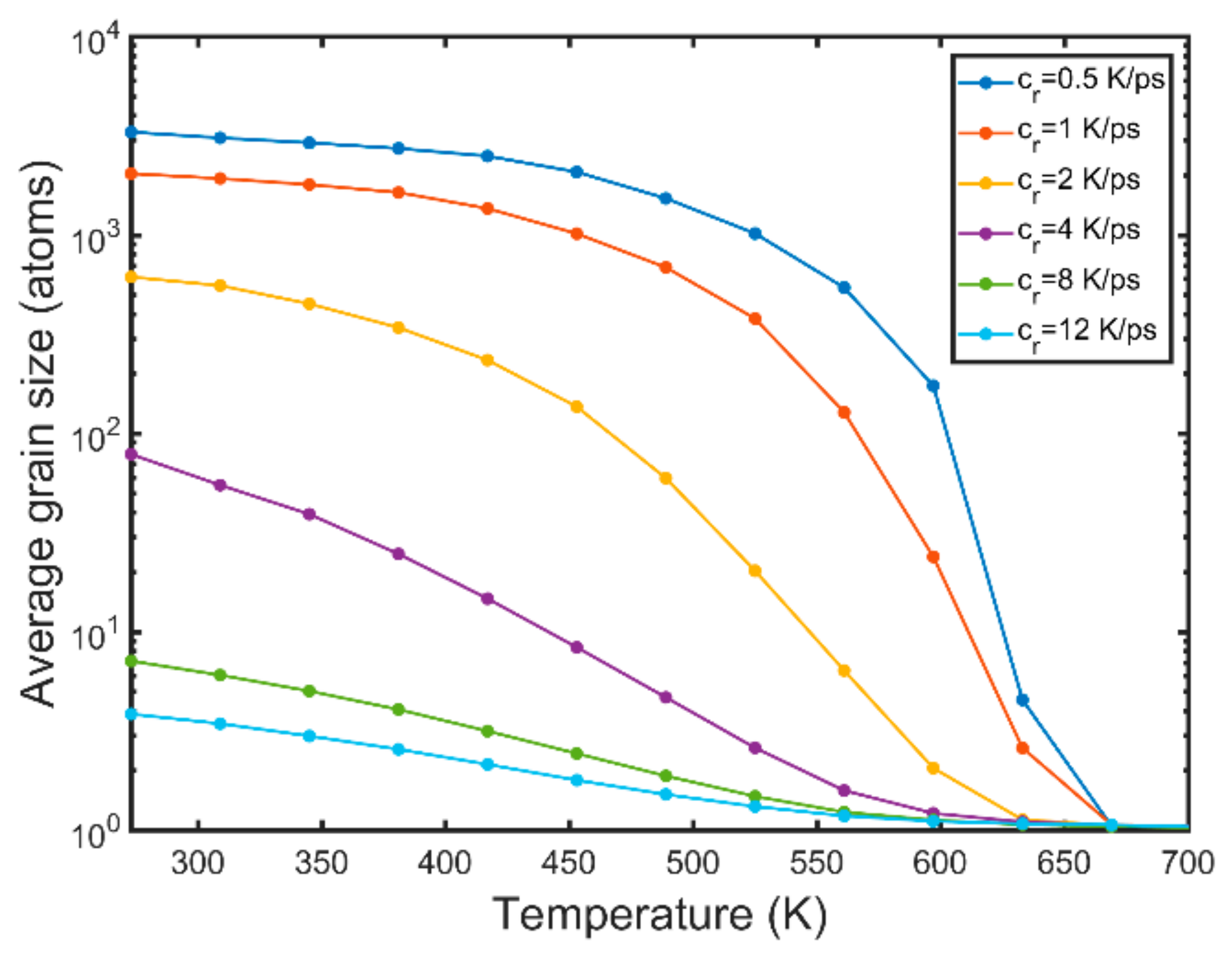
- High cooling rates favour the formation of a large number of small grains while low cooling rates generate fewer but larger grains. The FCC/HCP phase is dominant for low cooling rates while the amorphous phase is favoured by rapid quenching.
- The average grain size at the end of quenching drops parabolically with the cooling rate.
- Kinetic energy rises sharply in the vicinity of the newly formed grains while the melt kinetic energy remains significantly lower. This is because of the exothermic nature of the solidification process which is accompanied by the release of the latent heat of crystallisation.
- The solid phase temperature diverges from the linear cooling curve. The magnitude of the observed divergence is non-monotonous and dependent on the nucleation and growth rates. The concurrent nucleation of multiple grains increases significantly the instant temperature of the solid phase when the FCC/HCP phase is dominant.
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hall, E.O. The Deformation and Ageing of Mild Steel: III Discussion of Results. Proc. Phys. Soc. Sect. B 1951, 64, 747–753. [Google Scholar] [CrossRef]
- Petch, N.J. The cleavage strength of polycrystals. J. Iron Steel Inst. 1953, 174, 25–28. [Google Scholar]
- Jackson, K.A. Kinetic Processes: Crystal Growth, Diffusion, and Phase Transformations in Materials; John Wiley & Sons: Hoboken, NJ, USA, 2006. [Google Scholar]
- Iqbal, N.; van Dijk, N.H.; Offerman, S.E.; Moret, M.P.; Katgerman, L.; Kearley, G.J. Real-time observation of grain nucleation and growth during solidification of aluminium alloys. Acta Mater. 2005, 53, 2875–2880. [Google Scholar] [CrossRef]
- Volmer, M.; Weber, A. Keimbildung in übersättigten Gebilden. Z. Phys. Chem. 1926, 119, 277–301. [Google Scholar] [CrossRef]
- Miller, R.C.; Anderson, R.J.; Kassner, J.L.; Hagen, D.E. Homogeneous nucleation rate measurements for water over a wide range of temperature and nucleation rate. J. Chem. Phys. 1983, 78, 3204–3211. [Google Scholar] [CrossRef]
- Brus, D.; Ždímal, V.; Uchtmann, H. Homogeneous nucleation rate measurements in supersaturated water vapor II. J. Chem. Phys. 2009, 131, 074507. [Google Scholar] [CrossRef] [PubMed]
- Russo, J.; Tanaka, H. The microscopic pathway to crystallization in supercooled liquids. Sci. Rep. 2012, 2, 505. [Google Scholar] [CrossRef] [PubMed]
- Karthika, S.; Radhakrishnan, T.K.; Kalaichelvi, P. A Review of Classical and Nonclassical Nucleation Theories. Cryst. Growth Des. 2016, 16, 6663–6681. [Google Scholar] [CrossRef]
- Del Vitto, A.; Pacchioni, G.; Delbecq, F.; Sautet, P. Au atoms and dimers on the MgO (100) surface: A DFT study of nucleation at defects. J. Phys. Chem. B 2005, 109, 8040–8048. [Google Scholar] [CrossRef] [PubMed]
- Gähler, F.; Benkert, K. Atomistic Simulations on Scalar and Vector Computers. In High Performance Computing on Vector Systems; Springer: Berlin/Heidelberg, Germany, 2006; pp. 173–186. [Google Scholar]
- Kumar, M.; Sasikumar, R.; Nair, P.K. Competition between nucleation and early growth of ferrite from austenite—Studies using cellular automaton simulations. Acta Mater. 1998, 46, 6291–6303. [Google Scholar] [CrossRef]
- Nie, Y.; Liang, C.; Cha, P.-R.; Colombo, L.; Wallace, R.M.; Cho, K. A kinetic Monte Carlo simulation method of van der Waals epitaxy for atomistic nucleation-growth processes of transition metal dichalcogenides. Sci. Rep. 2017, 7, 2977. [Google Scholar] [CrossRef] [PubMed]
- Qin, R.S.; Bhadeshia, H.K.D.H. Phase-field model study of the effect of interface anisotropy on the crystal morphological evolution of cubic metals. Acta Mater. 2009, 57, 2210–2216. [Google Scholar] [CrossRef]
- Papanikolaou, M.; Salonitis, K. Fractal roughness effects on nanoscale grinding. Appl. Surf. Sci. 2019, 467–468, 309–319. [Google Scholar] [CrossRef]
- Papanikolaou, M.; Salonitis, K. Contact stiffness effects on nanoscale high-speed grinding: A molecular dynamics approach. Appl. Surf. Sci. 2019, 493, 212–224. [Google Scholar] [CrossRef]
- Fu, T.; Peng, X.; Zhao, Y.; Sun, R.; Weng, S.; Feng, C.; Wang, Z. Molecular dynamics simulation of TiN (001) thin films under indentation. Ceram. Int. 2015, 41, 14078–14086. [Google Scholar] [CrossRef]
- Povarnitsyn, M.E.; Fokin, V.B.; Levashov, P.R.; Itina, T.E. Molecular dynamics simulation of subpicosecond double-pulse laser ablation of metals. Phys. Rev. B 2015, 92, 174104. [Google Scholar] [CrossRef]
- Tian, Z.-A.; Liu, R.-S.; Liu, H.-R.; Zheng, C.-X.; Hou, Z.-Y.; Peng, P. Molecular dynamics simulation for cooling rate dependence of solidification microstructures of silver. J. Non-Cryst. Solids 2008, 354, 3705–3712. [Google Scholar] [CrossRef]
- Tian, Z.-A.; Liu, R.-S.; Zheng, C.-X.; Liu, H.-R.; Hou, Z.-Y.; Peng, P. Formation and Evolution of Metastable bcc Phase during Solidification of Liquid Ag: A Molecular Dynamics Simulation Study. J. Phys. Chem. A 2008, 112, 12326–12336. [Google Scholar] [CrossRef] [PubMed]
- Streitz, F.H.; Glosli, J.N.; Patel, M. V Beyond finite-size scaling in solidification simulations. Phys. Rev. Lett. 2006, 96, 225701. [Google Scholar] [CrossRef] [PubMed]
- Mahata, A.; Zaeem, M.A.; Baskes, M.I. Understanding homogeneous nucleation in solidification of aluminum by molecular dynamics simulations. Model. Simul. Mater. Sci. Eng. 2018, 26, 025007. [Google Scholar] [CrossRef]
- Hou, Z.Y.; Dong, K.J.; Tian, Z.A.; Liu, R.S.; Wang, Z.; Wang, J.G. Cooling rate dependence of solidification for liquid aluminium: A large-scale molecular dynamics simulation study. Phys. Chem. Chem. Phys. 2016, 18, 17461–17469. [Google Scholar] [CrossRef] [PubMed]
- Shibuta, Y.; Sakane, S.; Miyoshi, E.; Okita, S.; Takaki, T.; Ohno, M. Heterogeneity in homogeneous nucleation from billion-atom molecular dynamics simulation of solidification of pure metal. Nat. Commun. 2017, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Finnis, M.W.; Sinclair, J.E. A simple empirical N-body potential for transition metals. Philos. Mag. A 1984, 50, 45–55. [Google Scholar] [CrossRef]
- Shibuta, Y.; Suzuki, T. Melting and solidification point of fcc-metal nanoparticles with respect to particle size: A molecular dynamics study. Chem. Phys. Lett. 2010, 498, 323–327. [Google Scholar] [CrossRef]
- Shibuta, Y.; Oguchi, K.; Ohno, M. Million-atom molecular dynamics simulation on spontaneous evolution of anisotropy in solid nucleus during solidification of iron. Scr. Mater. 2014, 86, 20–23. [Google Scholar] [CrossRef]
- Stefanescu, D.M. Science and Engineering of Casting Solidification; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Lutsko, J.F. How crystals form: A theory of nucleation pathways. Sci. Adv. 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- LAMMPS Molecular Dynamics Simulator. Available online: https://lammps.sandia.gov/ (accessed on 1 October 2019).
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO-the Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 2010, 18. [Google Scholar] [CrossRef]
- Honeycutt, J.D.; Andersen, H.C. Molecular dynamics study of melting and freezing of small Lennard-Jones clusters. J. Phys. Chem. 1987, 91, 4950–4963. [Google Scholar] [CrossRef]
- Stranski, I.N.; Totomanow, D. Keimbildungsgeschwindigkeit und Ostwaldsche Stufenregel. Z. Phys. Chem. 1933, 163, 399–408. [Google Scholar] [CrossRef]
- Rein ten Wolde, P.; Ruiz-Montero, M.J.; Frenkel, D. Numerical calculation of the rate of crystal nucleation in a Lennard-Jones system at moderate undercooling. J. Chem. Phys. 1996, 104, 9932–9947. [Google Scholar] [CrossRef]
- Lü, Y.J.; Chen, M.; Yang, H.; Yu, D.Q. Nucleation of Ni–Fe alloy near the spinodal. Acta Mater. 2008, 56, 4022–4027. [Google Scholar] [CrossRef]
- Schenk, T.; Holland-Moritz, D.; Simonet, V.; Bellissent, R.; Herlach, D.M. Icosahedral short-range order in deeply undercooled metallic melts. Phys. Rev. Lett. 2002, 89, 75507. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Tian, Z.; Liu, R.; Dong, K.; Yu, A. Formation mechanism of bulk nanocrystalline aluminium with multiply twinned grains by liquid quenching: A molecular dynamics simulation study. Comput. Mater. Sci. 2015, 99, 256–261. [Google Scholar] [CrossRef]
- Schumacher, P.; Greer, A.L.; Worth, J.; Evans, P.V.; Kearns, M.A.; Fisher, P.; Green, A.H. New studies of nucleation mechanisms in aluminium alloys: Implications for grain refinement practice. Mater. Sci. Technol. 1998, 14, 394–404. [Google Scholar] [CrossRef]
- Nowak, M.; Bolzoni, L.; Hari Babu, N. Grain refinement of Al–Si alloys by Nb–B inoculation. Part I: Concept development and effect on binary alloys. Mater. Des. 2015, 66, 366–375. [Google Scholar] [CrossRef]
- Sosso, G.C.; Chen, J.; Cox, S.J.; Fitzner, M.; Pedevilla, P.; Zen, A.; Michaelides, A. Crystal Nucleation in Liquids: Open Questions and Future Challenges in Molecular Dynamics Simulations. Chem. Rev. 2016, 116, 7078–7116. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Atoms | 1,000,188 |
| Timestep (fs) | 2 |
| Pressure (Pa) | 0 |
| Ensemble | NPT |
| Al-Mass (amu) | 26.98 |
| Cooling Rates (K/ps) | 0.5, 1, 2, 4, 8, 12 |
| Temperature Damping Parameter (ps) | 0.2 |
| Pressure Damping Parameter (ps) | 2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papanikolaou, M.; Salonitis, K.; Jolly, M.; Frank, M. Large-Scale Molecular Dynamics Simulations of Homogeneous Nucleation of Pure Aluminium. Metals 2019, 9, 1217. https://doi.org/10.3390/met9111217
Papanikolaou M, Salonitis K, Jolly M, Frank M. Large-Scale Molecular Dynamics Simulations of Homogeneous Nucleation of Pure Aluminium. Metals. 2019; 9(11):1217. https://doi.org/10.3390/met9111217
Chicago/Turabian StylePapanikolaou, Michail, Konstantinos Salonitis, Mark Jolly, and Michael Frank. 2019. "Large-Scale Molecular Dynamics Simulations of Homogeneous Nucleation of Pure Aluminium" Metals 9, no. 11: 1217. https://doi.org/10.3390/met9111217
APA StylePapanikolaou, M., Salonitis, K., Jolly, M., & Frank, M. (2019). Large-Scale Molecular Dynamics Simulations of Homogeneous Nucleation of Pure Aluminium. Metals, 9(11), 1217. https://doi.org/10.3390/met9111217