Atomistic Simulations of Pure Tin Based on a New Modified Embedded-Atom Method Interatomic Potential

Abstract
1. Introduction
2. Methods
2.1. Construction of a DFT Database
2.2. Optimization of Potential Parameters
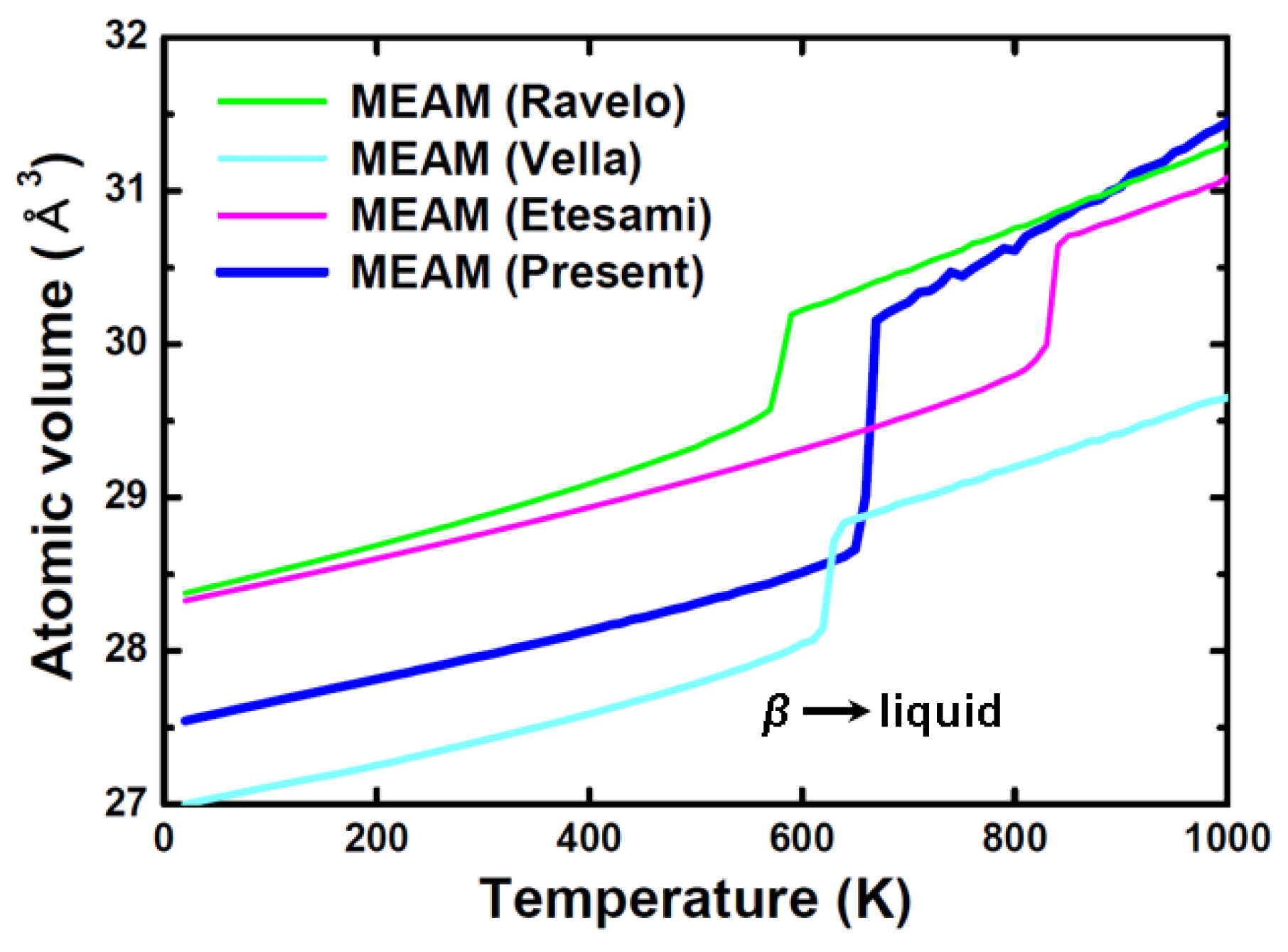
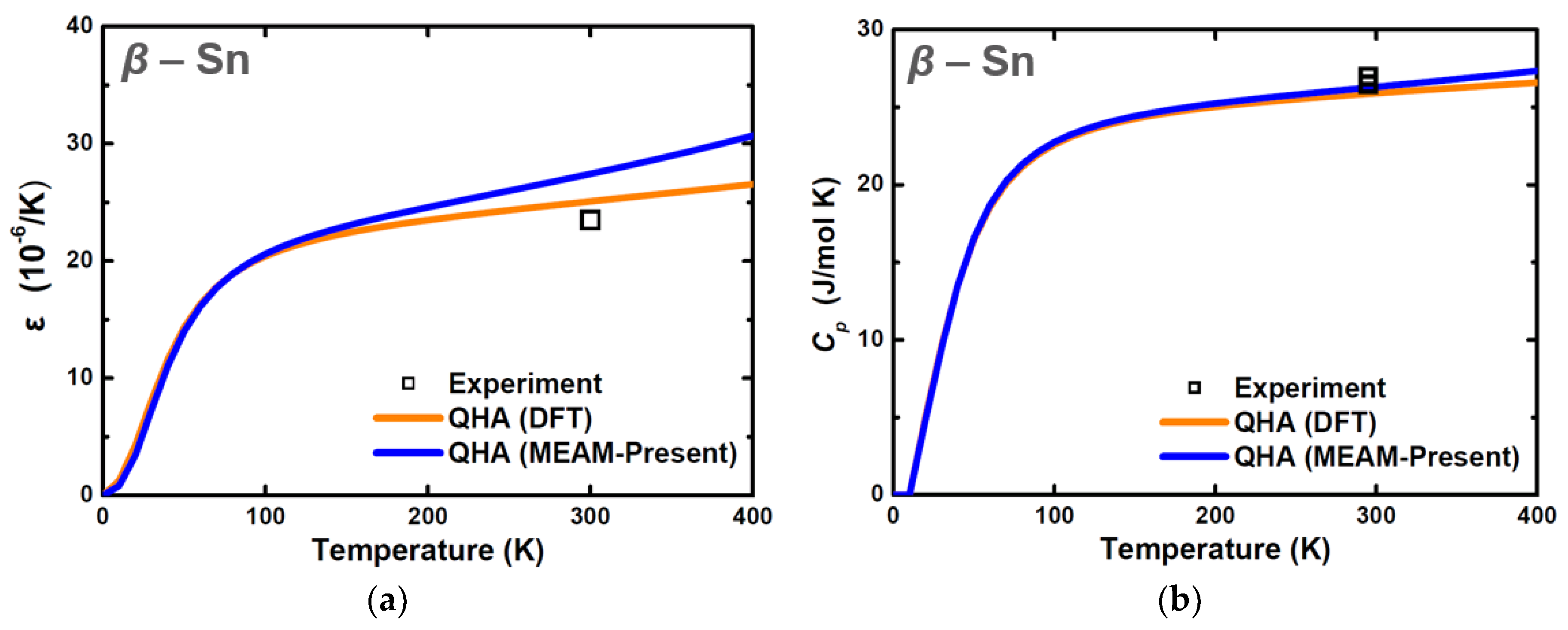
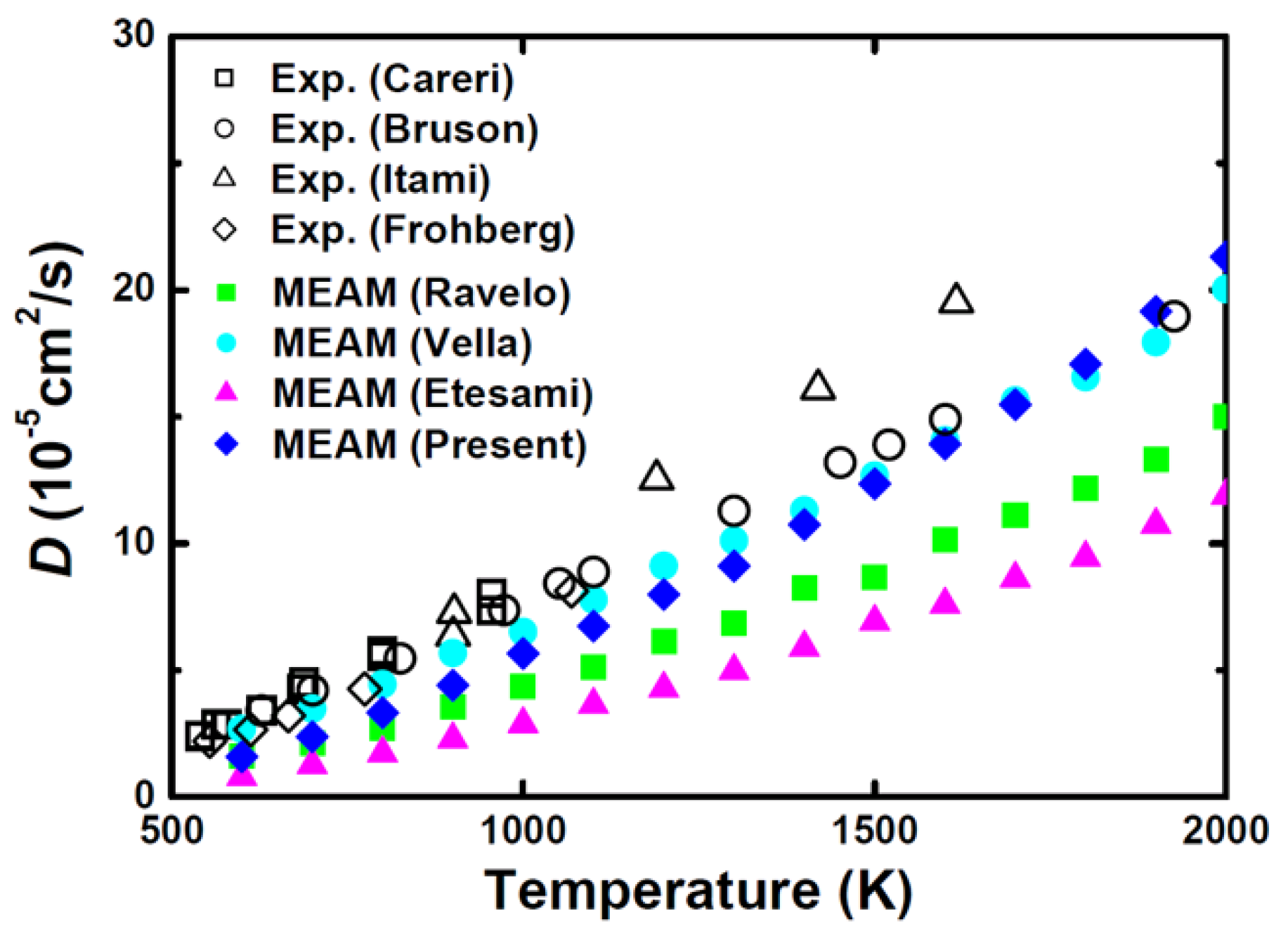
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Roshanghias, A.; Vrestal, J.; Yakymovych, A.; Richter, K.W.; Ipser, H. Sn-Ag-Cu nanosolders: Melting behavior and phase diagram prediction in the Sn-rich corner of the ternary system. Calphad 2015, 49, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Scrosati, B.; Garche, J. Lithium batteries: Status, prospects and future. J. Power Sources 2010, 195, 2419–2430. [Google Scholar] [CrossRef]
- Allain, J.P.; Ruzic, D.N.; Hendricks, M.R.D. He and Li sputtering of liquid eutectic Sn-Li. J. Nucl. Mater. 2001, 290–293, 33–37. [Google Scholar] [CrossRef]
- Coenen, J.W.; Temmerman, G.D.; Federici, G.; Philipps, V.; Sergienko, G.; Strohmayer, G.; Terra, A.; Unterberg, B.; Wegener, T.; Bekerom, D.C.M.V.D. Liquid metals as alternative solution for the power exhaust of future fusion devices: Status and perspective. Phys. Scr. 2014, 2014, 014037. [Google Scholar] [CrossRef]
- Ravelo, R.; Baskes, M. Equilibrium and Thermodynamic Properties of Grey, White, and Liquid Tin. Phys. Rev. Lett. 1997, 79, 2482–2485. [Google Scholar] [CrossRef]
- Masaki, T.; Aoki, H.; Munejiri, S.; Ishii, Y.; Itami, T. Effective pair interatomic potential and self-diffusion of molten tin. J. Non-Cryst. Solids 2002, 312–314, 191–195. [Google Scholar] [CrossRef]
- Itami, T.; Munejiri, S.; Masaki, T.; Aoki, H.; Ishii, Y.; Kamiyama, T.; Senda, Y.; Shimojo, F.; Hoshino, K. Structure of liquid Sn over a wide temperature range from neutron scattering experiments and first-principles molecular dynamics simulation: A comparison to liquid Pb. Phys. Rev. B 2003, 67, 064201. [Google Scholar] [CrossRef]
- Mouas, M.; Gasser, J.-G.; Hellal, S.; Grosdidier, B.; Makradi, A.; Belouettar, S. Diffusion and viscosity of liquid tin: Green-Kubo relationship-based calculations from molecular dynamics simulations. J. Chem. Phys. 2012, 136, 094501. [Google Scholar] [CrossRef] [PubMed]
- Sapozhnikov, F.A.; Ionov, G.V.; Dremov, V.V.; Soulard, L.; Durand, O. The Embedded Atom Model and large-scale MD simulation of tin under shock loading. J. Phys. Conf. Ser. 2014, 500, 032017. [Google Scholar] [CrossRef]
- Daw, M.S.; Baskes, M.I. Embedded-atom method: Derivation and application to impurities, surfaces, and other defects in metals. Phys. Rev. B 1984, 29, 6443–6453. [Google Scholar] [CrossRef]
- Baskes, M.I. Modified embedded-atom potentials for cubic materials and impurities. Phys. Rev. B 1992, 46, 2727–2742. [Google Scholar] [CrossRef]
- Vella, J.R.; Chen, M.; Stillinger, F.H.; Carter, E.A.; Debenedetti, P.G.; Panagiotopoulos, A.Z. Structural and dynamic properties of liquid tin from a new modified embedded-atom method force field. Phys. Rev. B 2017, 95, 064202. [Google Scholar] [CrossRef]
- Etesami, S.A.; Baskes, M.I.; Laradji, M.; Asadi, E. Thermodynamics of solid Sn and PbSn liquid mixtures using molecular dynamics simulations. Acta Mater. 2018, 161, 320–330. [Google Scholar] [CrossRef]
- Lee, B.-J.; Baskes, M.I. Second nearest-neighbor modified embedded-atom-method potential. Phys. Rev. B 2000, 62, 8564–8567. [Google Scholar] [CrossRef]
- Lee, B.-J.; Baskes, M.I.; Kim, H.; Cho, Y.K. Second nearest-neighbor modified embedded atom method potentials for bcc transition metals. Phys. Rev. B 2001, 64, 184102. [Google Scholar] [CrossRef]
- Lee, B.-J.; Ko, W.-S.; Kim, H.-K.; Kim, E.-H. The modified embedded-atom method interatomic potentials and recent progress in atomistic simulations. Calphad 2010, 34, 510–522. [Google Scholar] [CrossRef]
- Ercolessi, F.; Adams, J.B. Interatomic Potentials from First-Principles Calculations: The Force-Matching Method. Eur. Lett. 1994, 26, 583. [Google Scholar] [CrossRef]
- Mendelev, M.I.; Han, S.; Srolovitz, D.J.; Ackland, G.J.; Sun, D.Y.; Asta, M. Development of new interatomic potentials appropriate for crystalline and liquid iron. Philos. Mag. 2003, 83, 3977–3994. [Google Scholar] [CrossRef]
- Brommer, P.; Gähler, F. Potfit: Effective potentials from ab initio data. Model. Simul. Mater. Sci. Eng. 2007, 15, 295. [Google Scholar] [CrossRef]
- Ko, W.-S.; Grabowski, B.; Neugebauer, J. Development and application of a Ni-Ti interatomic potential with high predictive accuracy of the martensitic phase transition. Phys. Rev. B 2015. [Google Scholar] [CrossRef]
- Ko, W.-S.; Jeon, J.B. Interatomic potential that describes martensitic phase transformations in pure lithium. Comput. Mater. Sci. 2017, 129, 202–210. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initiomolecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Birch, F. Finite strain isotherm and velocities for single-crystal and polycrystalline NaCl at high pressures and 300°K. J. Geophys. Res. Solid Earth 1978, 83, 1257–1268. [Google Scholar] [CrossRef]
- Murnaghan, F.D. The Compressibility of Media under Extreme Pressures. Proc. Natl. Acad. Sci. USA 1944, 30, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Henkelman, G.; Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef]
- Togo, A.; Oba, F.; Tanaka, I. First-principles calculations of the ferroelastic transition between rutile-type and CaCl2-type SiO2 at high pressures. Phys. Rev. B 2008, 78, 134106. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. Evolution of crystal structures in metallic elements. Phys. Rev. B 2013, 87, 184104. [Google Scholar] [CrossRef]
- Wei, S.; Chou, M.Y. Ab initio calculation of force constants and full phonon dispersions. Phys. Rev. Lett. 1992, 69, 2799–2802. [Google Scholar] [CrossRef] [PubMed]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Liu, P.; Wang, S.; Li, D.; Li, Y.; Chen, X.-Q. Fast and Huge Anisotropic Diffusion of Cu (Ag) and Its Resistance on the Sn Self-diffusivity in Solid β-Sn. J. Mater. Sci. Technol. 2016, 32, 121–128. [Google Scholar] [CrossRef]
- Coston, C.; Nachtrieb, N.H. Self-Diffusion in Tin at High Pressure. J. Phys. Chem. 1964, 68, 2219–2229. [Google Scholar] [CrossRef]
- Kittel, C. Introduction to Solid State Physics, 8th ed.; Wiley: New York, NY, USA, 2005. [Google Scholar]
- Barrett, C.S.; Massalski, T.B. Structure of Metals; McGraw-Hill: New York, NY, USA, 1966. [Google Scholar]
- Brandes, E.A.; Brook, G.B. Smithells Metals Reference Book, 7th ed.; Butterworth-Heinemann: Oxford, UK, 1992. [Google Scholar]
- Ihm, J.; Cohen, M.L. Equilibrium properties and the phase transition of grey and white tin. Phys. Rev. B 1981, 23, 1576–1579. [Google Scholar] [CrossRef]
- Harrison, P.G. Chemistry of Tin; Blackie: Glasgow, Scotland, 1989. [Google Scholar]
- Barin, I.; Knacke, O.; Kubaschewski, O. Thermochemical Properties of Inorganic Substances; Springer-Verlag: Berlin, Germany, 1973. [Google Scholar]
- Morris, J.R.; Wang, C.Z.; Ho, K.M.; Chan, C.T. Melting line of aluminum from simulations of coexisting phases. Phys. Rev. B 1994, 49, 3109–3115. [Google Scholar] [CrossRef]
- Zhu, L.-F.; Grabowski, B.; Neugebauer, J. Efficient approach to compute melting properties fully from ab initio with application to Cu. Phys. Rev. B 2017, 96, 224202. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015. [Google Scholar] [CrossRef]
- Dean, J.A. Lange’s Handbook of Chemistry; McGraw-Hill: New York, NY, USA, 1985. [Google Scholar]
- Bruson, A.; Gerl, M. Diffusion coefficient of 113Sn, 124Sb, 110mAg, and 195Au in liquid Sn. Phys. Rev. B 1980, 21, 5447–5454. [Google Scholar] [CrossRef]
- Careri, G.; Paoletti, A.; Vicentini, M. Further experiments on liquid Indium and Tin self-diffusion. Il Nuovo Cimento 2008, 10, 1088. [Google Scholar] [CrossRef]
- Itami, T.; Aoki, H.; Kaneko, M.; Uchida, M.; Shisa, A.; Amano, S.; Odawara, O.; Masaki, T.; Oda, H.; Ooida, T.; et al. Diffusion of Liquid Metals and Alloys—The study of self-diffusion under microgravity in liquid Sn in the wide temperature range. Jpn. Soc. Micrograv. Appl. 1998, 15, 225. [Google Scholar]
- Frohberg, G.; Kraatz, K.H.; Weber, H. Diffusion and transport phenomena in liquids under microgravity. In Proceedings of the 6th European Symposium on Materials Sciences under Microgravity Conditions, Bordeaux, France, 2–5 December 1986; ESA: Paris, France. [Google Scholar]
- Available online: http://www.ctcms.nist.gov/potentials (accessed on 1 October 2018).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Stability | Temp. (K) | Strain (%) | k-Point | ||
|---|---|---|---|---|---|---|
| diamond cubic (α) | 64 | 0 | low T stable | 0, 300 | 0, H (±5, ±10) | 5 × 5 × 5 |
| 64 | 0 | low T stable | 0 | O (±5, ±10), M (±4, ±8) | 5 × 5 × 5 | |
| 63 | 1 | low T stable | 0, 300 | 0, H (±5, ±10) | 5 × 5 × 5 | |
| 62 | 2 | low T stable | 0 | 0 | 5 × 5 × 5 | |
| body-centered tetragonal (β) | 64 | 0 | high T stable | 0, 300 | 0, H (±5, ±10) | 5 × 5 × 5 |
| 64 | 0 | high T stable | 0 | O (±10), M (±7) | 5 × 5 × 5 | |
| 63 | 1 | high T stable | 0, 300 | 0, H (±5, ±10) | 5 × 5 × 5 | |
| 62 | 2 | high T stable | 0 | 0 | 5 × 5 × 5 | |
| fcc | 108 | 0 | hypothetical | 0 | 0 | 4 × 4 × 4 |
| bcc | 128 | 0 | hypothetical | 0 | 0 | 4 × 4 × 4 |
| hcp | 96 | 0 | hypothetical | 0 | 0 | 4 × 4 × 5 |
| liquid | 64 | 0 | stable | 400, 500, 600 | 0 | 5 × 5 × 5 |
| B | A | d | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3.05 | 3.480 | 0.6088 | 1.05 | 5.50 | 5.10 | 4.50 | 4.30 | 1.30 | 3.60 | −0.90 | 1.29 | 4.43 | 0.02 |
| Phase | Property | Exp. | DFT f | MEAM g,h [Ravelo] | MEAM g [Vella] | MEAM g [Etesami] | 2NN MEAM [This Work] |
|---|---|---|---|---|---|---|---|
| diamond cubic (α) | 3.140 a | 3.154 | 3.140 | 3.220 | 3.209 | 3.135 | |
| 6.483 b | 6.658 | 6.483 | 6.304 | 6.430 | 6.581 | ||
| 0.426 c | 0.358 | 0.422 | 0.442 | 0.436 | 0.406 | ||
| 0.691 c | - | 0.704 | 0.649 | 0.819 | 0.504 | ||
| 0.213 c | - | 0.281 | 0.339 | 0.244 | 0.357 | ||
| 0.426 c | - | 0.367 | 0.426 | 0.949 | 0.105 | ||
| - | 0.041 | 0.055 | 0.105 | 0.118 | 0.033 | ||
| - | 0.065 | 0.060 | 0.160 | 0.129 | 0.052 | ||
| - | 0.075 | 0.060 | 0.144 | 0.129 | 0.053 | ||
| - | 0.063 | 0.059 | 0.160 | 0.128 | 0.050 | ||
| body-centeredtetragonal (β) | 3.10 d | 3.113 | 3.085 | 3.115 | 3.091 | 3.102 | |
| 5.831 b | 5.938 | 5.920 | 5.682 | 5.914 | 5.859 | ||
| 3.184 b | 3.224 | 3.235 | 3.334 | 3.237 | 3.206 | ||
| 0.546 b | 0.543 | 0.546 | 0.587 | 0.547 | 0.547 | ||
| 0.570 c | 0.479 | 0.645 | 0.656 | 0.647 | 0.571 | ||
| 0.734 c | - | 1.093 | 1.233 | 1.329 | 0.897 | ||
| 0.599 c | - | 0.625 | 0.343 | 0.469 | 0.467 | ||
| 0.391 c | - | 0.244 | 0.540 | 0.166 | 0.369 | ||
| 0.907 c | - | 1.396 | 0.592 | 1.571 | 0.937 | ||
| 0.220 c | - | 0.007 | 0.042 | 0.012 | 0.079 | ||
| 0.239 c | - | 0.225 | 0.215 | 0.281 | 0.106 | ||
| - | 0.734 | 1.112 | 1.545 | 1.617 | 0.848 | ||
| - | 0.082 | - | - | - | 0.235 | ||
| - | 0.290 | 0.257 | 0.268 | 0.439 | 0.291 | ||
| 1.089 e | 0.816 | - | - | - | 1.083 | ||
| 1.111 e | 1.023 | 1.369 | 1.813 | 2.057 | 1.139 | ||
| - | 378 | 725 | 813 | 1375 | 345 | ||
| - | 359 | 889 | 792 | 1300 | 393 |
| Property | Exp. | MEAM d,e [Ravelo] | MEAM d [Vella] | MEAM d [Etesami] | MEAM [This Work] |
|---|---|---|---|---|---|
| (300 K) | 23.5 a | 23.0 | 20.3 | 19.3 | 18.6 |
| (295 K) | 26.5 b | 27.2 | 26.6 | 26.5 | 26.1 |
| 505 c | - | 435 | 502 | 368 | |
| 7.0 c | - | 4.2 | 4.1 | 3.1 | |
| 2.3 c | - | 2.6 | 2.5 | 4.4 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ko, W.-S.; Kim, D.-H.; Kwon, Y.-J.; Lee, M.H. Atomistic Simulations of Pure Tin Based on a New Modified Embedded-Atom Method Interatomic Potential. Metals 2018, 8, 900. https://doi.org/10.3390/met8110900
Ko W-S, Kim D-H, Kwon Y-J, Lee MH. Atomistic Simulations of Pure Tin Based on a New Modified Embedded-Atom Method Interatomic Potential. Metals. 2018; 8(11):900. https://doi.org/10.3390/met8110900
Chicago/Turabian StyleKo, Won-Seok, Dong-Hyun Kim, Yong-Jai Kwon, and Min Hyung Lee. 2018. "Atomistic Simulations of Pure Tin Based on a New Modified Embedded-Atom Method Interatomic Potential" Metals 8, no. 11: 900. https://doi.org/10.3390/met8110900
APA StyleKo, W.-S., Kim, D.-H., Kwon, Y.-J., & Lee, M. H. (2018). Atomistic Simulations of Pure Tin Based on a New Modified Embedded-Atom Method Interatomic Potential. Metals, 8(11), 900. https://doi.org/10.3390/met8110900