1. Introduction
The wet chemical decoating of metal cutting tools allows for the removal of abraded or faulty coatings and prepares them for subsequent reuse. Therefore, a complete and substrate-sensitive technique is necessary to do this. Previous studies [
1,
2,
3,
4] have indicated that a mixture of 3.0 mol/L ammonia, 4.7 mol/L hydrogen peroxide, and 0.1 mol/L citric acid at the appropriate process parameters has led to the best results with respect to these requirements. In former investigations, commercially available indexable inserts, which consisted of WC-MX-Co cemented carbides coated with hard materials, were treated. These inserts involved TiAlN-PVD, TiAlTaN-PVD, and TiB
2-CVD coatings as well as a TiN-bonding layer in each case to improve the wear resistance of the cutting tools. Because cemented carbides principally represent a composite material of tungsten carbide and cobalt [
5,
6] and because there is inevitable degradation of the substrate during the decoating procedure, the spent etchants contain considerable amounts of Co and W. Furthermore, these solutions exhibit other metal ions, such as Ti or Ta, which originate from the dissolved hard coatings. The great number of different substances in the used stripping agents requires processing to reduce the hazardous components, especially Co.
Spent stripping solutions, which have arisen from the previously mentioned studies [
1,
2,
3,
4], have chemical compositions that are not common by-products of any known processes. Therefore, these liquids represent an exceptional residue for which a novel approach must first be established in the laboratory, because no standardised reconditioning step is currently available. Hence, this study was performed to develop an environmentally friendly and low-waste technique to process these alkaline solutions comprising spent ammonia, hydrogen peroxide, and citric acid and containing Co, Ti, and other elements. However, another question that has arisen is whether the production of a secondary, solid raw material is possible within the process; such a material would constitute a useful basis for the recovery of valuable components, such as Ti.
As mentioned before, one of the main components of these liquids is citric acid. This colourless, odourless, and crystalline solid has the molecular formula C
6H
8O
7. Its monohydrate and anhydrous compounds melt at 100 °C and 153 °C, respectively. Citric acid is a weak acid that dissociates stepwise in water, as given in Equations (1)–(4), whereby the distribution of the citric ions is dependent on the dissociation constants and the pH value. H
3AOH represents citric acid, and A corresponds with C
6H
4O
6 [
7,
8,
9,
10].
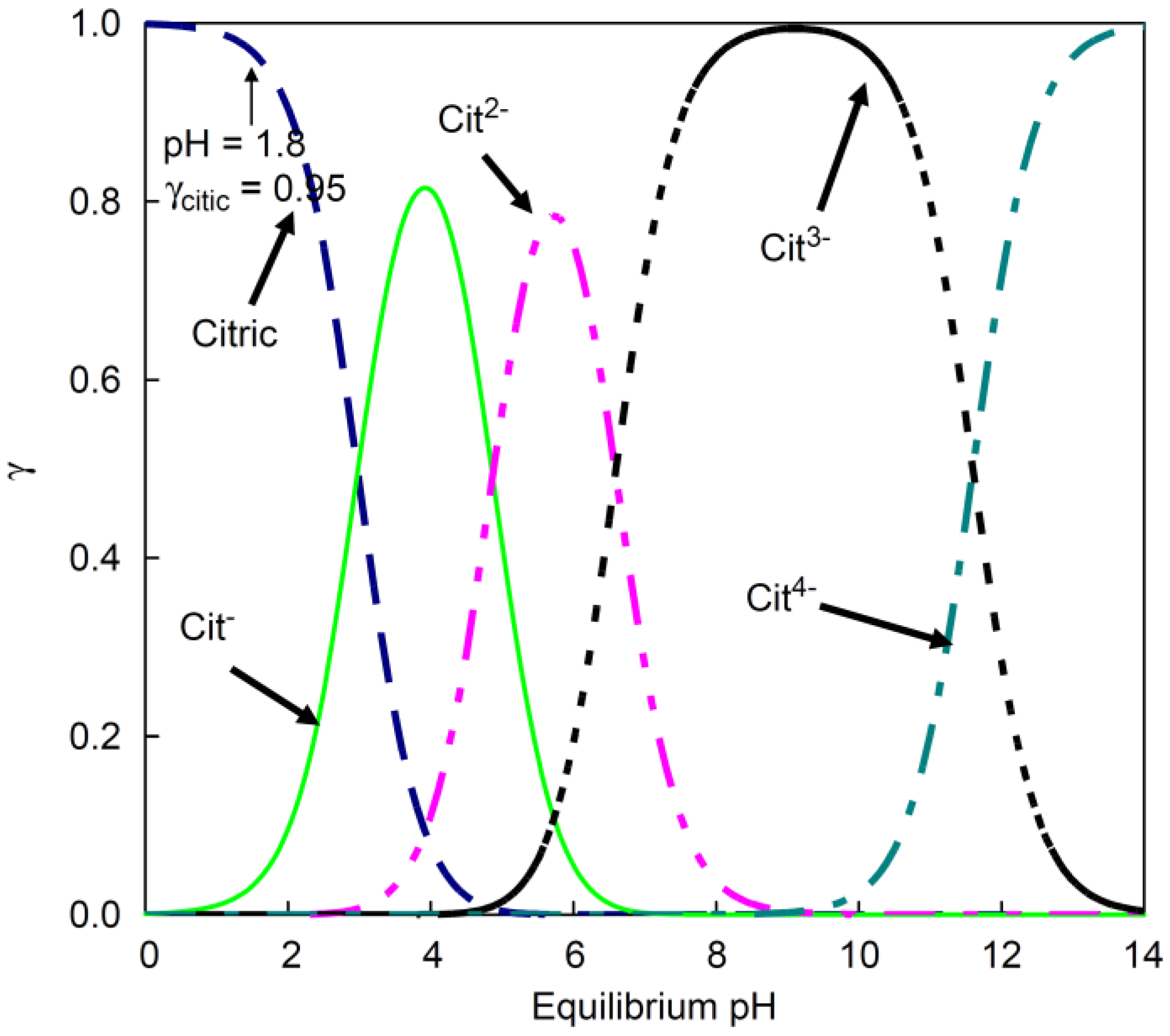
According to Al-Khaldi et al. (2007),
Figure 1 represents the distribution of citric acid ions depending on the equilibrium pH value. As can be seen, AOH
3− respectively Cit
3− constitutes the predominant citric acid species in the alkaline milieu, which is the focus of the current study [
10].
Citric acid forms thermodynamically stable chelate complexes with various metal ions, such as Co, Ni, Cu, Fe, and W. In doing so, bonds between the carboxyl or hydroxyl groups and the metal ion occur. As stated by Wyrzykowsky and Chmurzyński (2010), Co
2+, Ni
2+, Mn
2+, or Zn
2+ form 1:1 complexes at a pH of 6 at 25 °C, because the equilibrium lies at Cit
2−, which can also be seen in
Figure 1. In some cases, more than one citric acid molecule can be involved in this reaction. Co and citrate ions, for example, can form different complex species in aqueous solutions, such as [Co(CitH)], [Co(Cit)]
−, [Co(Cit)
2]
4−, [Co(CitH
-1)]
2−, or [Co
2(CitH
-1)
2]
4−. Their distribution depends on the pH value, as well as the metal/ligand ratio in the liquid. This formation of stable complexes impedes the precipitation of the solid metal-containing compounds, leading to certain challenges with respect to the removal of the hazardous components [
9,
10,
11,
12].
This problem can be solved by the addition of a Ca source and its dissolution into the spent stripping solutions. In the presence of calcium ions, citric acid combines, for example, to form CaHCit, CaH
2Cit
+, or Ca
3Cit
2. The latter exhibits low solubility in water, which decreases with the rising temperature [
13,
14].
Because the citrate is mainly present as a trivalent Cit
3− in the alkaline spent stripping solutions, which have been processed in this study, and tricalcium dicitrate is a known calcium salt of citric acid, the following chemical equation was assumed to take place during the precipitation:
Taking advantage of these circumstances enables the processing of the spent stripping agents and the diminution of the metal ions.
2. Materials and Methods
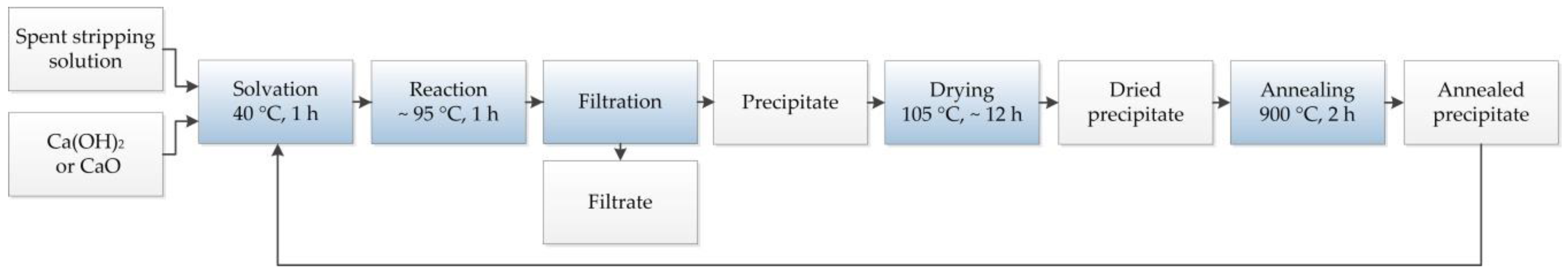
Our suggested method for processing alkaline, metal-containing spent stripping solutions in a laboratory model, therefore, is based on the precipitation of an insoluble compound through the use of a Ca source. The following flow chart, given in
Figure 2, shows the steps of the developed treatment.
First, Ca(OH)
2 or CaO (Carl Roth GmbH + Co. KG, Karlsruhe, Germany) was added to the liquid as a Ca
2+ source in a certain excess. While heating the suspension, the citrate reacted with the dissolved calcium ions to form a poorly soluble calcium citrate compound, which precipitated as a white, voluminous solid. The solid reaction product was separated from the solution by filtration. After drying, the filter cake had to be annealed in air for 2 h at 900 °C. This temperature appeared appropriate for the thermal treatment, because Ca citrates decompose stepwise in CaO through the emission of CO
2, H
2O, and other volatile constituents. Furthermore, the excess of the undissolved Ca(OH)
2 reacted to CaO.
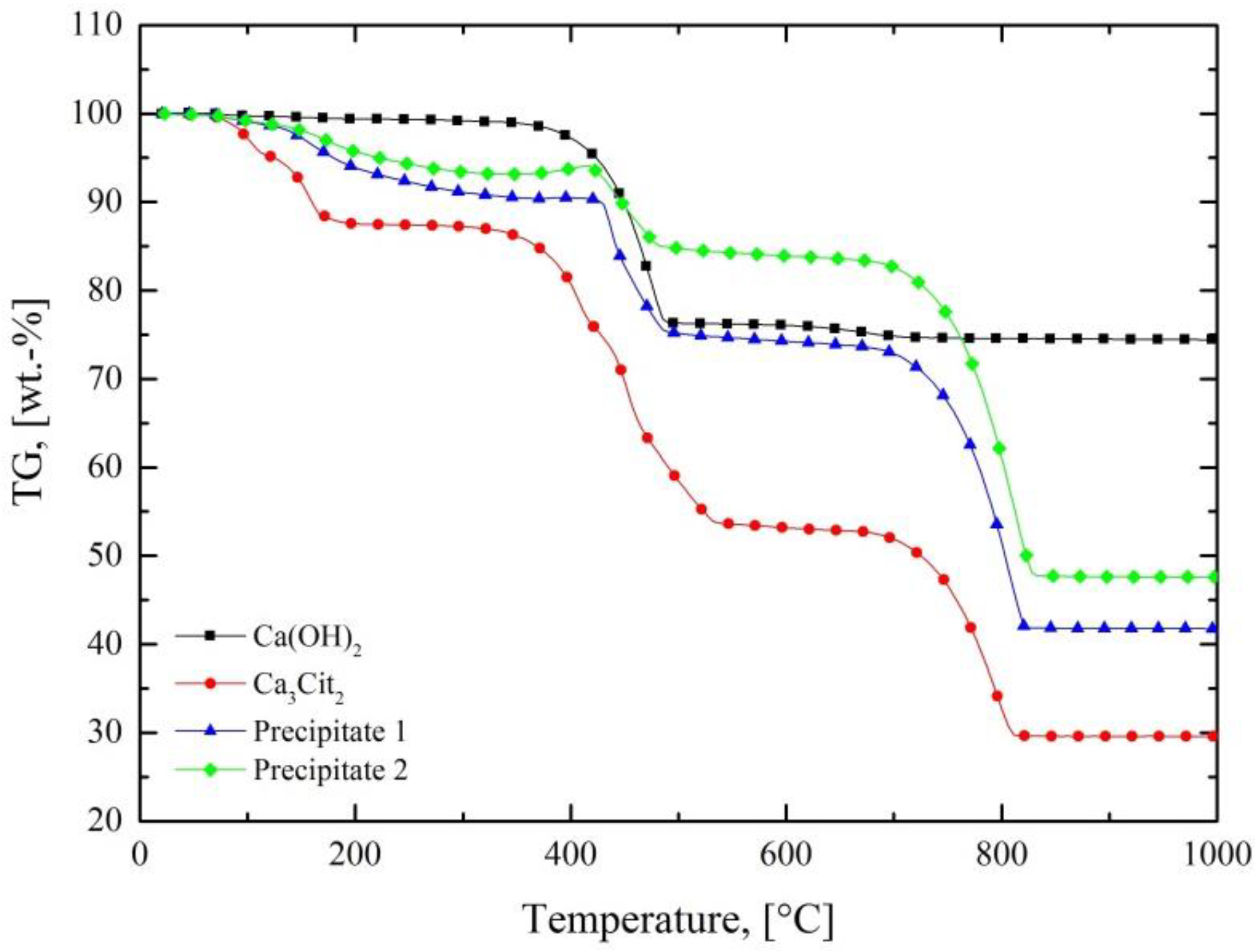
Figure 3 depicts the thermogravimetric analyses of pure Ca(OH)
2 and Ca
3Cit
2, as well as of two precipitates from preliminary investigations, in which Ca(OH)
2 served as the precipitating agent. The graph shows the weight losses that occurred in the course of heating these substances to 1000 °C in air. A stepwise decomposition of the materials took place until a constant weight was reached. The product of the thermal procedure, calcium oxide, represented a suitable input material, as a Ca
2+ source for a renewed use as a precipitating agent for citrates in stripping solutions. This enabled a closed-loop circulation of the solid residue in the form of a low-waste process, in which the losses needed to be compensated for with a fresh material. Aside from the diminution of the citrates, a decline of the metal ion concentrations of the solution occurred, which resulted in an enrichment of certain components in the precipitate. Hence, this method represents an effective preliminary step for the recovery of concentrated compounds from such a solid.
Two different series of tests were performed. Series 1 was meant to provide information regarding the effectiveness of the developed technique and to clarify if the reuse of the thermally treated residue was possible without adding a fresh Ca source. Series 2 was applied with CaO as the starting material in constant excess.
The experimental procedure was based on the processing method depicted in
Figure 2. First, 200 mL of the starting solution was heated to 40 °C while being stirred in a beaker. After reaching this temperature, the addition of a fresh Ca source occurred in the first experiment of each series of the thermally treated precipitate for the subsequent tests. The temperature remained constant for 1 h. Meanwhile, the soluble amount of the solid dissolved. During the ensuing boiling of the suspension for another hour, a calcium citrate compound precipitated in the form of a white and voluminous solid. As required, the evaporated water was compensated for with the addition of deionized water. After cooling the suspension to the ambient temperature, the suspended particles were separated by filtration. The drying of the filter cake took place at 105 °C until its weight remained constant. Then, it was annealed in air for 2 h at 900 °C. The thermally treated residue served as a precipitating agent for the removal of the citrates in the following tests. Depending on the aim of the series, fresh Ca(OH)
2 (series 1) or CaO (series 2) was added to the solid. Deionized water was added to the filtrate, which originated from the solid–liquid separation, until the solution reached 200 mL. The solution was analysed with ion chromatography (IC, Ion Chromatography System ICS-2000, Dionex Corporation, Sunnyvale, CA, USA) and inductively coupled plasma mass spectrometry (ICP-MS, Agilent 7500 CE, Agilent Technologies, Santa Clara, CA, USA) to measure the citrate and the metal content of the liquid, respectively.
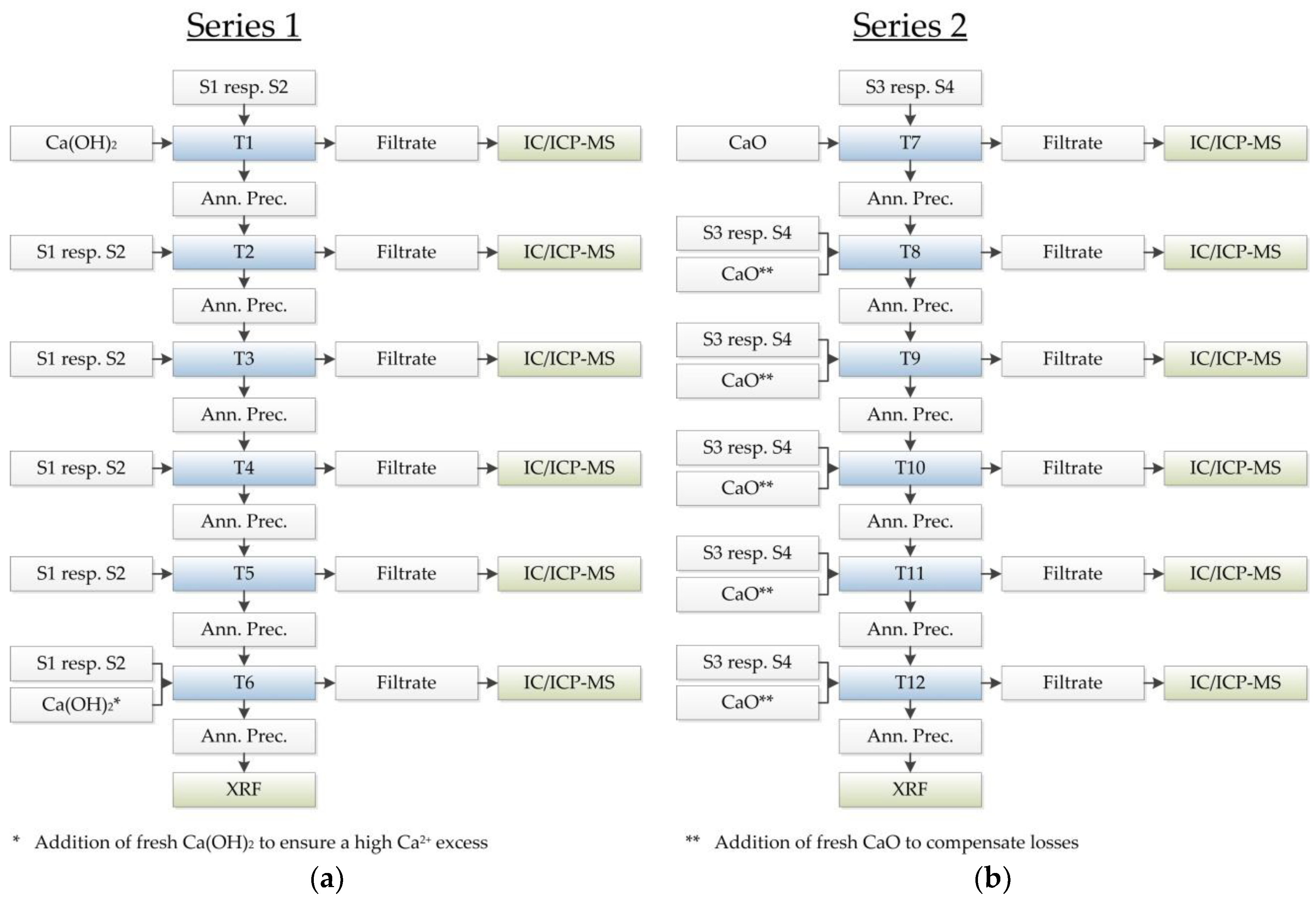
In series 1 (test designations T1–T6,
Figure 4a), 200 mL of two different spent stripping solutions, S1 and S2, were processed by adding Ca
2+ ions in the form of Ca(OH)
2 in triple excess in relation to the citrate concentration present in each solution (T1). The resulting thermally treated residues served as starting materials for the respective, subsequent tests (T2–T5), which were used without the addition of a fresh Ca source. In the last test of the first series (T6), Ca(OH)
2 was added to the remaining solid, which ensured a high excess of Ca
2+ ions. Over the course of the first 5 tests (T1–T5) of series 1, the number of Ca
2+ ions declined because of losses based on, for example, the significant Ca concentrations in the processed solutions, the filtration, and the handling. The annealed residues, which resulted from the last tests (T6), were analysed by X-ray fluorescence (XRF, XRF WDXRF Axios Panalytical, Malvern Panalytical, Malvern, UK; Almelo, Netherlands) to determine its chemical composition and draw conclusions concerning the metal content of the solid.
Series 2 comprised of 6 experiments (test designations T7–T12,
Figure 4b) with 200 mL of two different initial solutions, S3 and S4, and was meant to provide information regarding whether the use of CaO as a precipitation agent was possible. After each test (T7–T11), the Ca losses were compensated for with the addition of fresh calcium oxide, which theoretically led to an approximately constant threefold excess of Ca
2+ ions over the course of the tests. Therefore, the simplifying assumption implicated that the solid resulting from the previous precipitation only consisted of CaO after the annealing step. The thermally treated solid residue of the last tests (T12) were analysed by XRF, as given in
Section 3.
Initial Solutions
Table 1 summarises the results of the IC and ICP-MS analyses of the 4 different, transparent, and solid-free initial solutions, which were treated in the course of this study. This investigation was performed with a focus on the dissolved metal ions Al, Co, Nb, Ta, Ti, and W, which represent the most important constituents of the applied spent stripping solutions. The liquids S1 and S2 were processed in series 1 (T1–T6), whereas S3 and S4 were examined in series 2 (T7–T12). The pH values of S1, S2, S3, and S4 corresponded with 10.3, 10.4, 10.0, and 9.6, respectively.
3. Results
Table 2 shows the multiple of the stoichiometrically required amount of Ca
2+, calculated by Equation (5), in relation to the citrate concentration present in each spent stripping agent. In the first tests of each series (T1 and T7), a fresh Ca source was applied as a precipitating agent. In series 1, using S1 and S2 as the initial solutions, the thermally treated residues were reused in the subsequent tests without the need of the further addition of a Ca source, as mentioned before. The relative amount declined from T1 to T5 because of losses concerning, for example, the dissolved Ca in the filtrates or the handling. In T5 for the processing of S1, the number of Ca
2+ ions fell under the stoichiometrically required quantity, so it can be hypothesised that it is not possible to remove all the present citrates. In the last tests (T6) of series 1, fresh Ca(OH)
2 was added to the solid for the precipitation, which led to a 5.63- and 5.56-fold excess of Ca
2+ in S1 and S2, respectively, which should have improved the effectiveness of the process to a greater extent. In series 2 (T7–T12), the Ca losses over the course of the experiments were compensated for with the addition of fresh CaO, which resulted in a theoretically constant threefold excess.
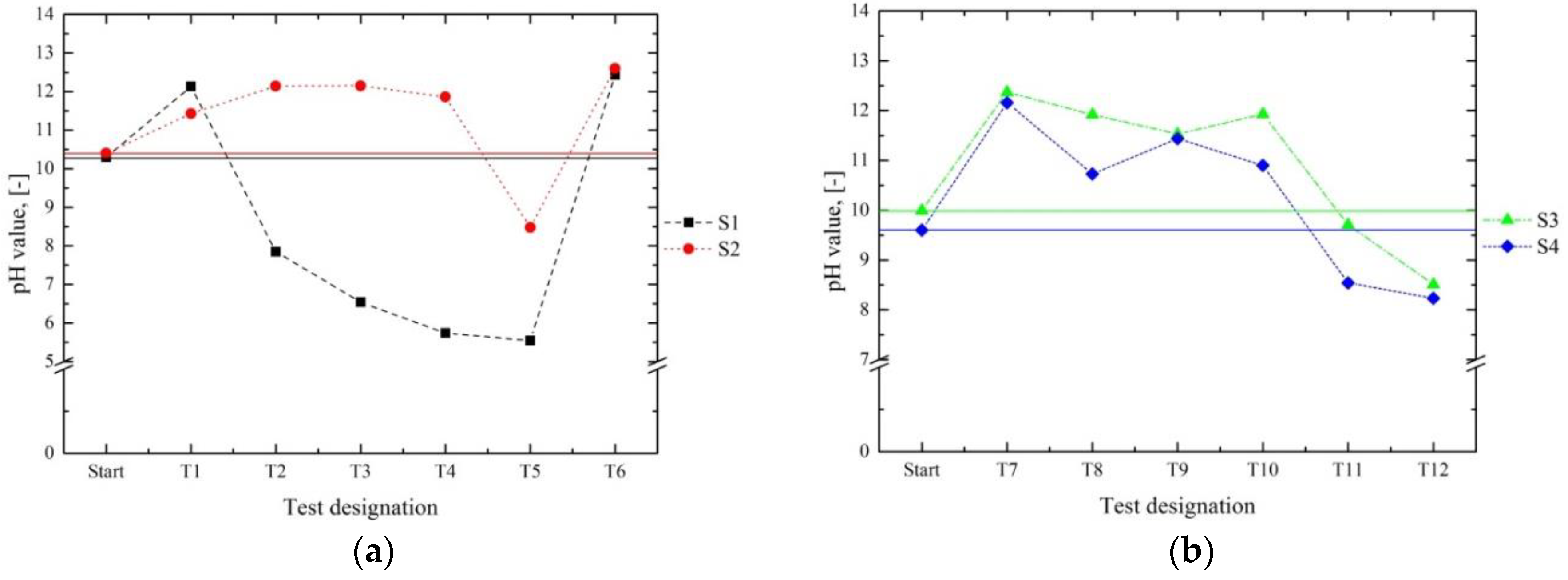
The following graphs in
Figure 5 compare the pH value before the experiment (start) with the pH value after the removal of the solid and the cooling of the filtrate of series 1 (
Figure 5a) and 2 (
Figure 5b). The pH of the filtrates resulting from the processing of S1 (
Figure 5a) surpassed the starting value in T1 and T6 only; in T2–T5, it declined constantly until the lowest point of 5.6 was reached in T5. All the final pH values of S2 (
Figure 5a) were in the alkaline range. In S2, the lowest pH was also reached in T5 and corresponded to about 8.5. The pH decreased over the course of the tests due to the inevitable losses of the solid. It was possible to counteract this trend by adding a fresh Ca source, as shown in T6. Based on the compensation for the losses in series 2 (
Figure 5b), the pH of the filtrates should have been more alkaline than the initial solutions S3 and S4. This assumption can only be confirmed for T7–T10. The reason for the lower pH values in T11 and T12 is discussed in the analysis of the XRF results below.
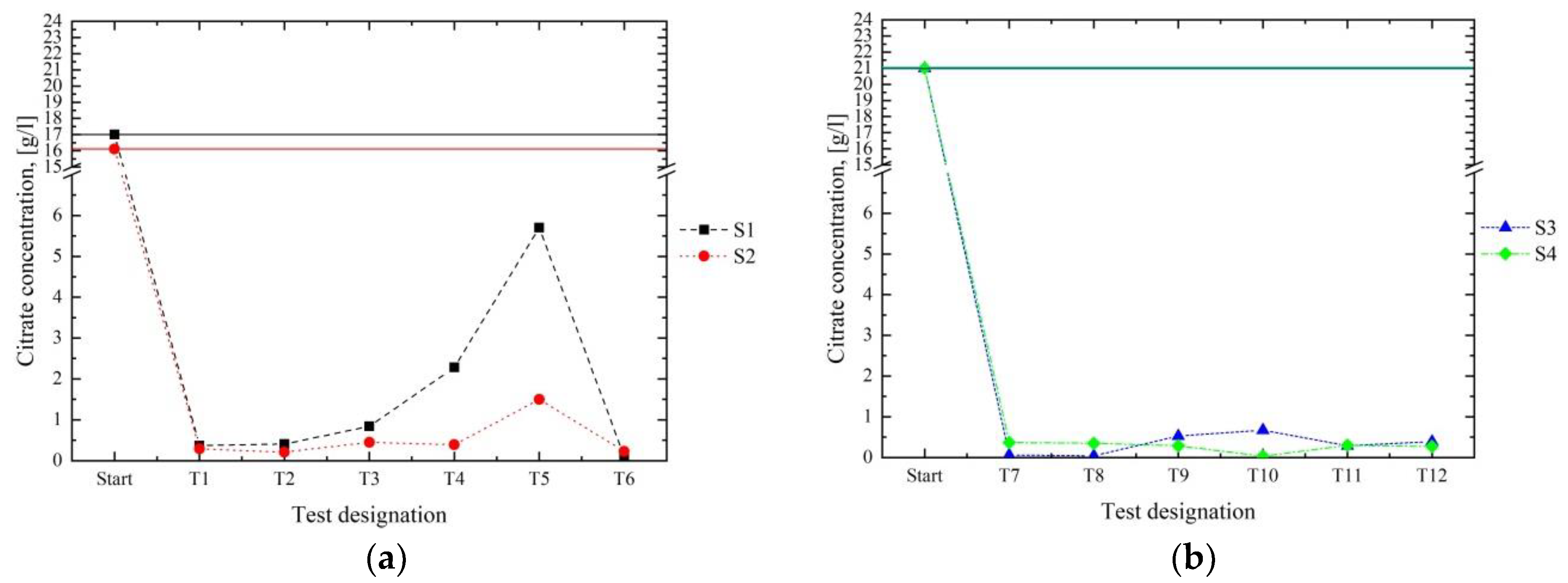
Figure 6 compares the citrate concentrations of the four initial solutions to the resulting filtrates from series 1 (
Figure 6a) and 2 (
Figure 6b). By analysing, for example, the outcome of T1 (
Figure 6a) and T7 (
Figure 6b), it can be concluded that a successful diminution of the citrates was achieved by the use of Ca(OH)
2 (series 1) and CaO (series 2), because 97.8% and 98.2% of the citrates in S1 and S2 of series 1, respectively, and 99.7% and 98.2% of the present citrates in S3 and S4 of series 2, respectively, were removed in the first step (
Table 3). Furthermore, we can conclude that the reuse of the thermally treated precipitate, according to the proposed process in
Figure 2, is possible. The effectiveness of the citrate reduction was associated with the presence of the Ca
2+ ions and the pH value. Hence, the contents of the citrates in series 1 (
Figure 6a) increased during the tests, because the amounts of the provided Ca
2+ and the pH declined, as given in
Table 2 and
Figure 5, especially in the T5 for the processing of S1 and S2. The low citrate concentrations of the series 2 filtrates (
Figure 6b) showed that the continuous addition of fresh CaO resulted in a satisfying reduction of the citrates, because the citrate concentrations constantly remained below 1 g/L. The comparison of the citrate contents and the pH values allowed us to conclude that the best results were achieved in an alkaline milieu (pH value > 8).
During the precipitation of the calcium citrates, a simultaneous diminution of most other dissolved metal ions occurred. As an example,
Table 3 shows the concentrations of the metal ions in the filtrates after the first tests of each series (T1 and T7). Furthermore,
Table 3 highlights the reduction in the concentrations in relation to the initial values, given in
Table 1. As can be seen, the contents of most of the metal ions, except for W in series 1, were significantly decreased because of the sufficient excess of the added Ca source. For example, a diminution of 96.4% and 99.2% was observed for Co in S1 and S2 in series 1, respectively, and a decrease of 98.8% and 97.2% occurred in S3 and S4 in series 2, respectively, which is of particular importance for this hazardous element.
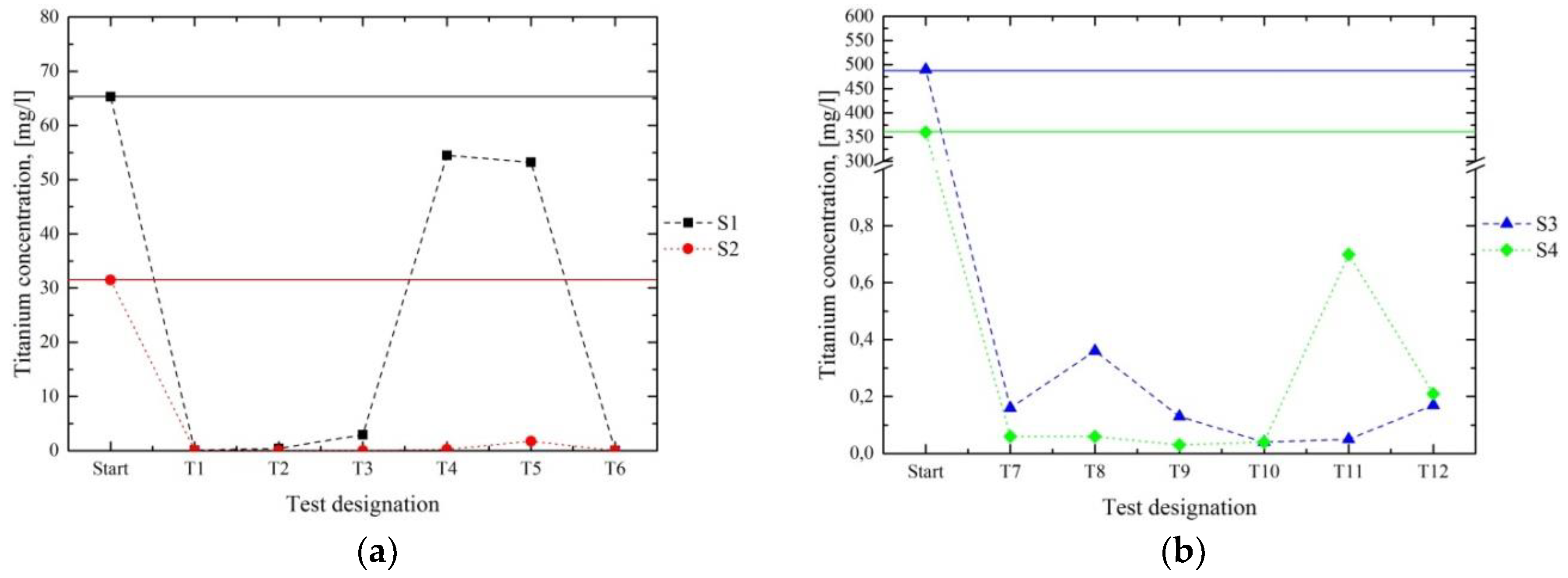
Figure 7,
Figure 8,
Figure 9,
Figure 10 and
Figure 11 compare the initial value (start,
Table 1) of an ion in the spent stripping solution to its concentration in the treated filtrates (T1–T6 and T7–T12). The values of the filtrates of T1 and T7 of each figure correspond with the values given in
Table 3.
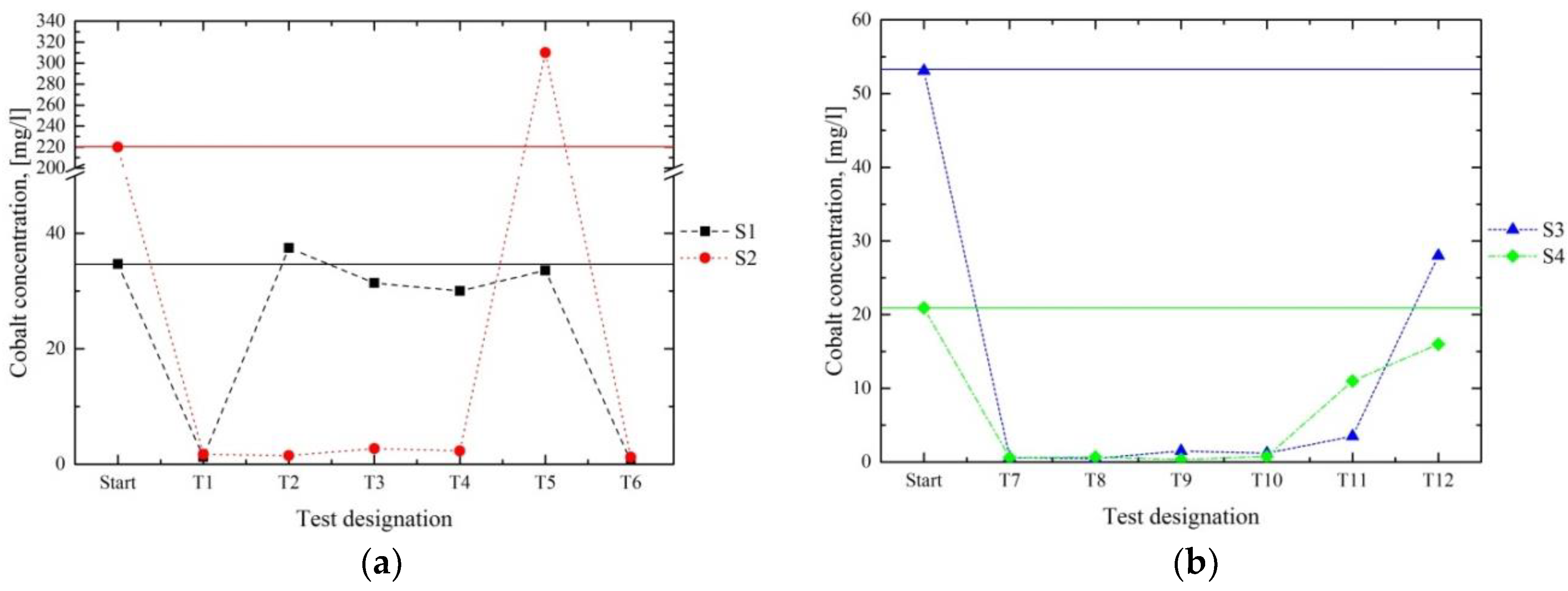
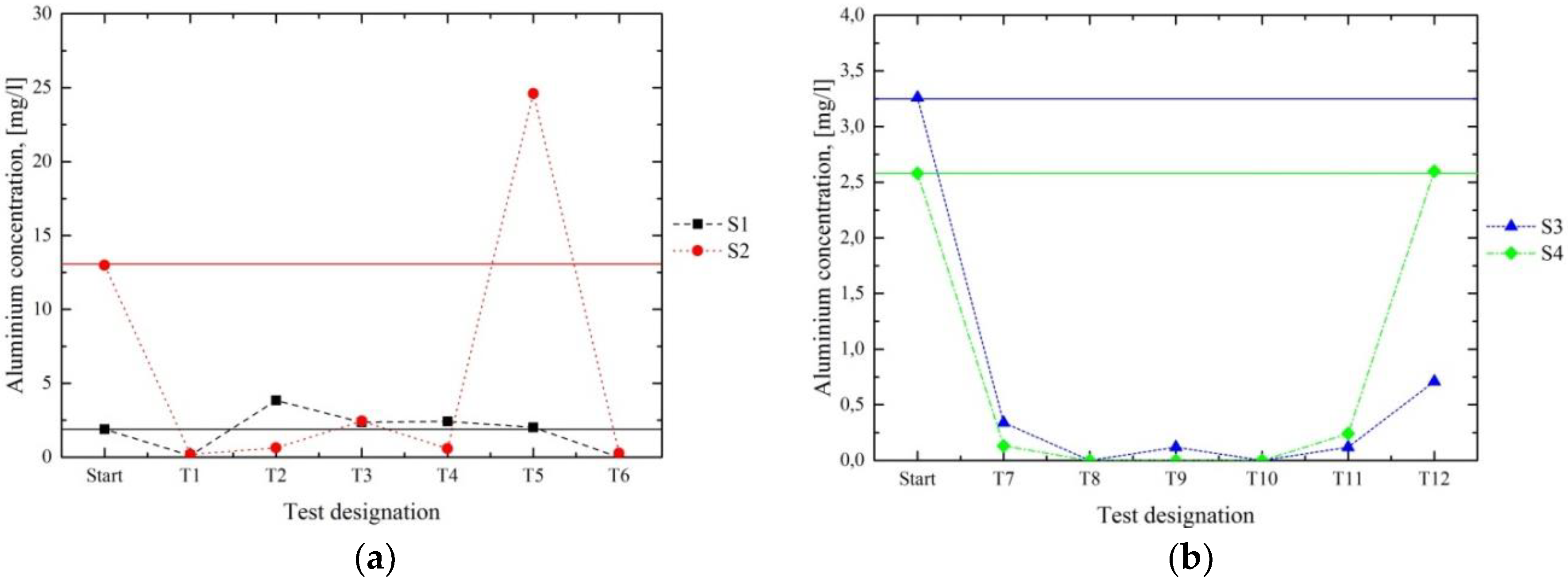
Figure 7 and
Figure 8 present the results for the Co and Al contents of the filtrates. In series 1 (a), the best results were achieved in the first and the last tests (T1 and T6) for both S1 and S2. Throughout the further processing of S1 in T2–T5, hardly any reduction of Co and Al occurred. During the processing of S2 in series 1, most of the Co and Al contents were removed, except for in T5. The results of series 2 (b) show satisfying results in T7–T10. These results indicated that the separation of Co and Al occurred most successfully in a strongly alkaline milieu at pH 10–12. In principle, the metal concentrations in the filtrate—which exceeded the initial contents in the initial solutions, such as in T5 in the processing of S2 for Co and Al—may be explained by the dissolution of an already-separated metal compound in the reused precipitate.
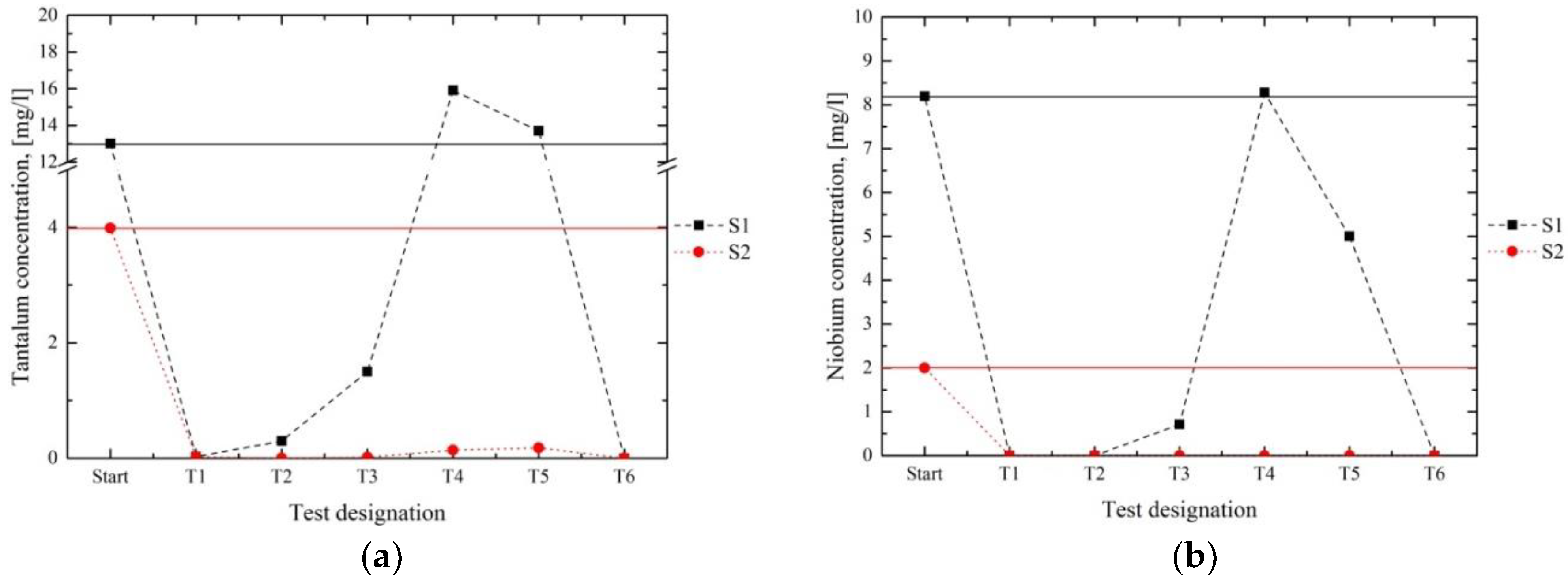
The graphs, given in
Figure 9 and
Figure 10a, present the removal of Ti and Ta compared with the initial concentrations. The evaluation of the graphs indicated that in series 1 (
Figure 9a,
Figure 10a) the best results were achieved in tests T1 and T6, whereas the amount of both elements increased in T2–T5. Series 2 exhibited a satisfying reduction of Ti (
Figure 9b) and Ta in each test run. Because the concentrations of Ta in the solutions of T7–T12 were located below the analytical limit, no graph is presented for those tests. Regarding the influence of the pH value, we have concluded that a satisfying diminution of both elements is possible in the alkaline range (pH value > 8).
Figure 10b displays the Nb concentrations before and after the tests in series 1. As seen in
Table 1, the amount of Nb in S3 and S4 was located below the analytical limit; hence, the results of series 2 are not presented here. For series 1, no Nb was detected in T1, T2, and T6 for S1. In T3–T5 only a partial diminution was achieved. In the course of the treatment of S2, no Nb was detected in any filtrate. Thus, a pH value > 8 led to the successful reduction of Nb.
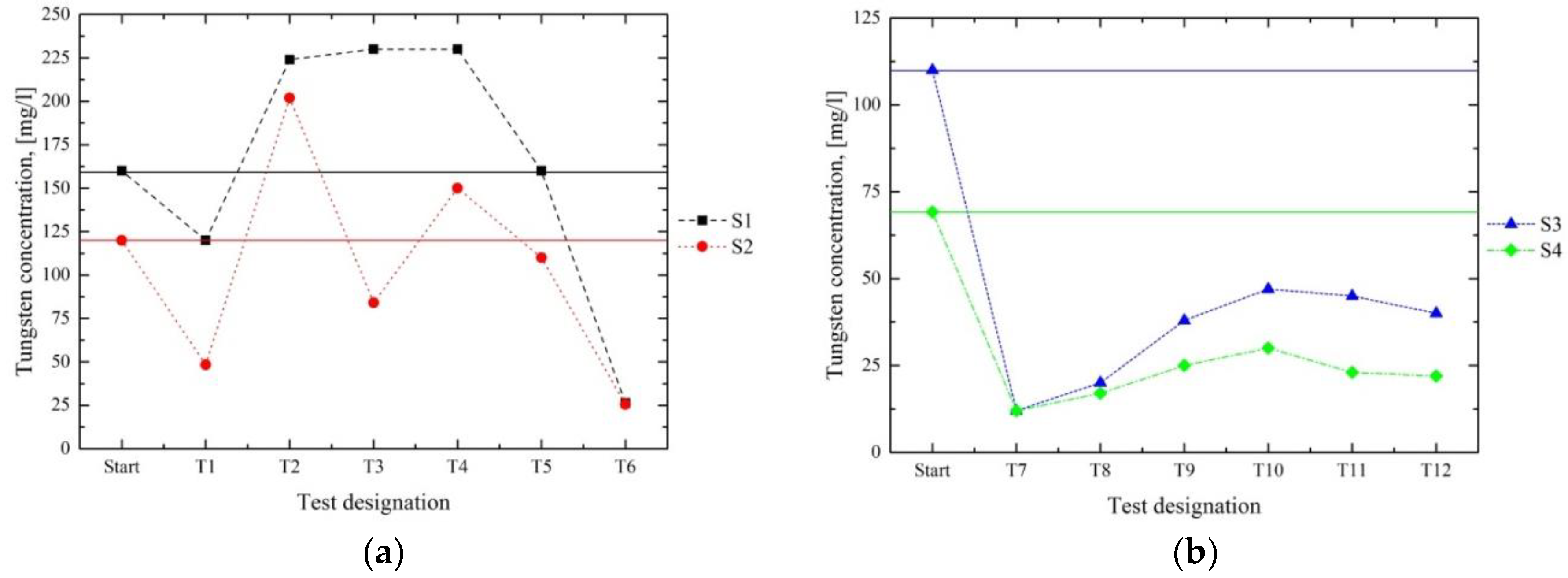
Figure 11 shows the W content of the filtrates after the tests and compares them with the initial values. The concentrations in series 1 (
Figure 11a) were only diminished significantly in T1 and T6. With respect to series 2 (
Figure 11b), the lowest contents of W were reached in the first test T7. These results indicated that it is possible to remove certain amounts of W; nevertheless, this element tended to remain dissolved, which implied that no complete precipitation can be achieved.
Table 4 represents the main components and trace elements of the annealed residues after series 1 and 2, which were analysed by XRF. All four solids comprised calcium oxide as a main component, with 92.9, 88.3, 65.4, and 72.7 wt.%, respectively. CaO was formed by annealing the filter cake at 900 °C in air via stepwise decomposition through the separation of H
2O, CO
2, and other volatile components. Because the solid was subsequently heated up to 1000 °C prior to the XRF analysis, an ignition loss occurred based on the removal of other volatile substances. Concerning the most important compounds formed by the substrate and coating elements, TiO
2 and Al
2O
3 were especially noteworthy. The content of Al
2O
3 accounted for 0.07, 0.28, 0.40, and 0.41 wt.% in the residues of S1, S2, S3, and S4, respectively. A clear difference between the two series can be seen regarding the TiO
2 contents, with 1.03 and 0.81 wt.% in S1 and S2 of series 1, respectively, and 21.0 and 17.0 wt.% in S3 and S4 of series 2, respectively. This can be explained by the enhanced removal of Ti in series 2 due to the theoretically constant excess of Ca
2+ ions and the higher Ti concentration in the initial solutions, as given in
Table 1. Other elements, such as Co, W, Ta, and Nb separated to certain extents from the liquid, but their enrichment in the solid merely remained in the ppm range.
4. Discussion
The results of this study allowed us to conclude that a removal of citrates from spent stripping solutions based on the proposed method, stated in
Figure 2, is possible. The findings of series 1 indicated that the separation rate can be enhanced by the addition of a fresh Ca compound. Hence, a refreshing of the annealed solid, for example, with Ca(OH)
2 or CaO appears reasonable. This presumption was confirmed by the outcome of series 2, because the reduction of the pH values in both of the final tests had no negative influence on the removal of the citrates. This follows the fact that the supply of Ca
2+ ions due to the addition of a fresh Ca source was sufficient for the formation of calcium citrate.
Parallel to the citrate removal over the course of precipitation, a diminution of various metal ions also occurred. As shown in
Table 3, the removal of a high proportion of most of the dissolved ions was observed, especially with a sufficient excess of a Ca source. The decisive factor for this diminution was the pH value. A decline of the pH to 8 (series 2) had no negative influence on the separation rate of the citrates; nevertheless, it led to an increase of several metal ions in the filtrates, as shown by the trend of Co in series 2, as shown in
Figure 7. The decrease of the pH in T11 and T12 of series 2 (
Figure 5b) can be explained by the aforementioned separation of the metal ions, such as Ti. Therefore, the annealed residue not only consisted of CaO, but also of a certain amount of other metal oxides, such as TiO
2. Because an accumulation of these oxides occurred in series 2, the decreasing amount of CaO and the subsequent decline of the pH value became especially apparent in the last tests T11 and T12. This was confirmed by the XRF analyses given in
Table 4, because the annealed solid arising from the processing of S3 only consisted of 65.4% CaO; the remaining part comprised various other oxides. As a consequence of this continuous enrichment, the aspired threefold excess from the addition of a fresh Ca source was not reached in the real system. Hence, the theoretical excess resembled the real excess at the beginning of series 2 but declined in further tests. This circumstance could be counteracted by analysing the residue after every test, which would give a better understanding of the oxidic content. The required amount of input material for the XRF measurement needed to be compensated for by adding fresh CaO in the precipitating agent, which constituted a source of error in the subsequent experiments. Because the necessary amount of solid for the analysis approximately corresponds with the mass of the annealed solid for processing 200 mL of the spent stripping solution, this analysis procedure seems to be disadvantageous.
The results of series 1 and 2 for Co and Al (
Figure 7 and
Figure 8) indicated that a successful removal of these elements is possible, especially in an alkaline medium. The best results were achieved at pH values around 12. For further relevant constituents, such as Ti, Ta, and Nb (
Figure 9 and
Figure 10), high separation rates were obtained as well, even at a comparatively lower pH value of > 8.
The removal of W (
Figure 11) presented a certain challenge, because the solubility of this element strongly depends on the pH value. The results of series 1 indicated that a high excess of Ca
2+ ions and the resulting pH values of 12.4 and 12.6 in T6 led to a reduction rate of 83.6% and 78.8% in S3 and S4, respectively. Consequently, no satisfying separation of W, just a certain diminution, was achieved. Nevertheless, these results allowed us to conclude that, at least, the removal of a high proportion of W occurs in a strongly alkaline milieu (pH value > 12).
With respect to the composition of the annealed solids (
Table 4) resulting from series 1 (S1 and S2), only a slight enrichment of the separated elements occurred. The content of 6800 ppm W in the annealed precipitate of S1 was attributed to the considerable concentration of 160 mg/L W in the initial solution (
Table 1). In contrast, the residue of S2 contained 25,430 ppm Co as a result of the high Co concentration of 220 mg/L in the spent stripping solution. Due to the enhancement in series 2 regarding the addition of a fresh Ca source, an enrichment of 21.0 and 17.0 wt.% TiO
2 was achieved. This provides a useful starting base for further investigations concerning the recovery of valuable components. The optimised procedure in series 2, furthermore, led to higher concentrations of W (14,300 and 6510 ppm in S3 and S4, respectively) and Co (11,100 and 4720 ppm in S3 and S4, respectively) in the annealed solid.

 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}