
Structural, Elastic, Electronic, Magnetic, and Half-Metallic Properties of Full-Heusler Compounds Fe2LiZ (Z = Ge and Si): A First-Principles Study


Abstract
1. Introduction
2. Computational Details
3. Results and Discussion
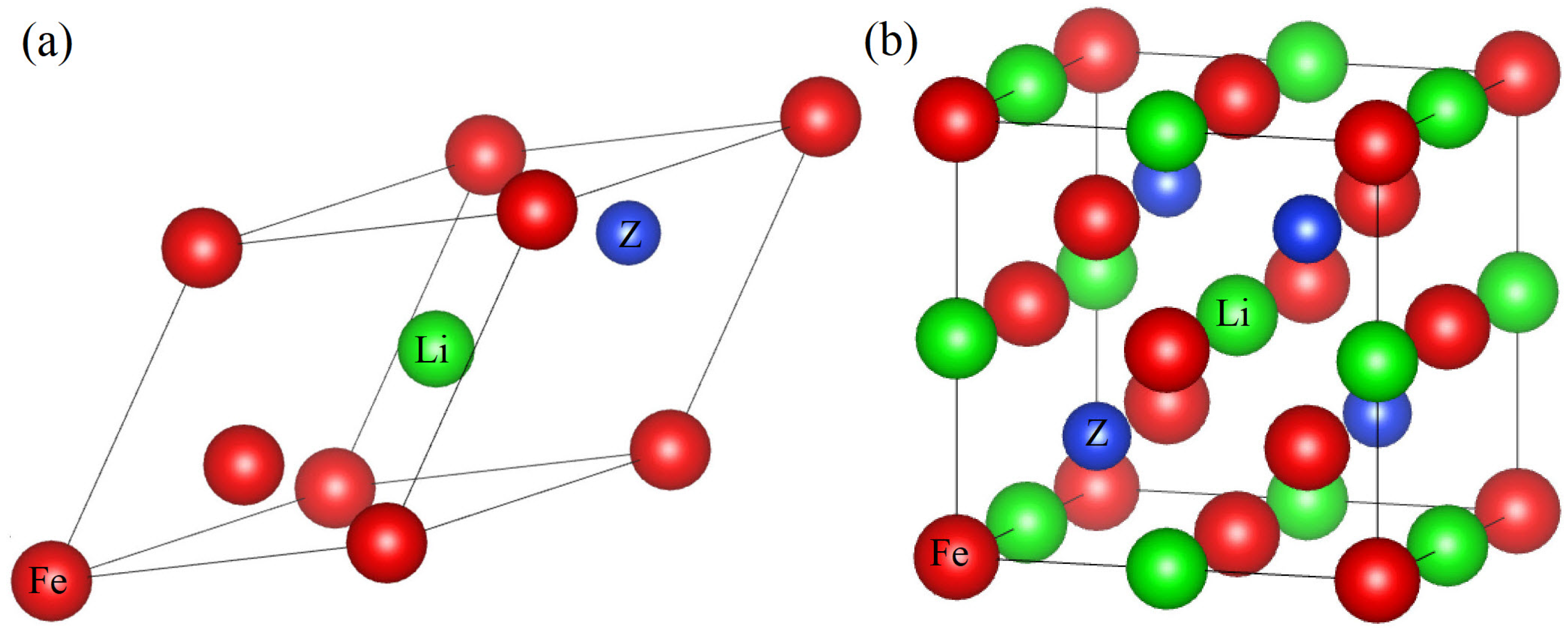
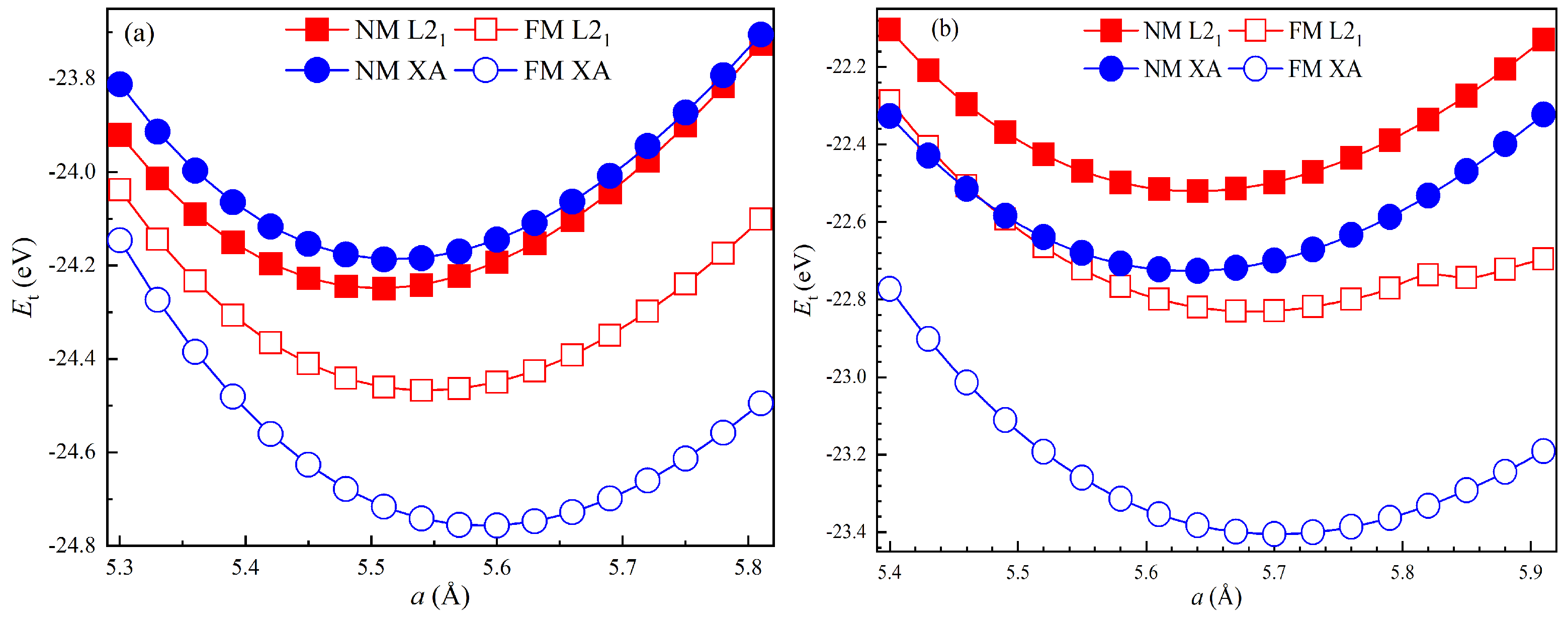
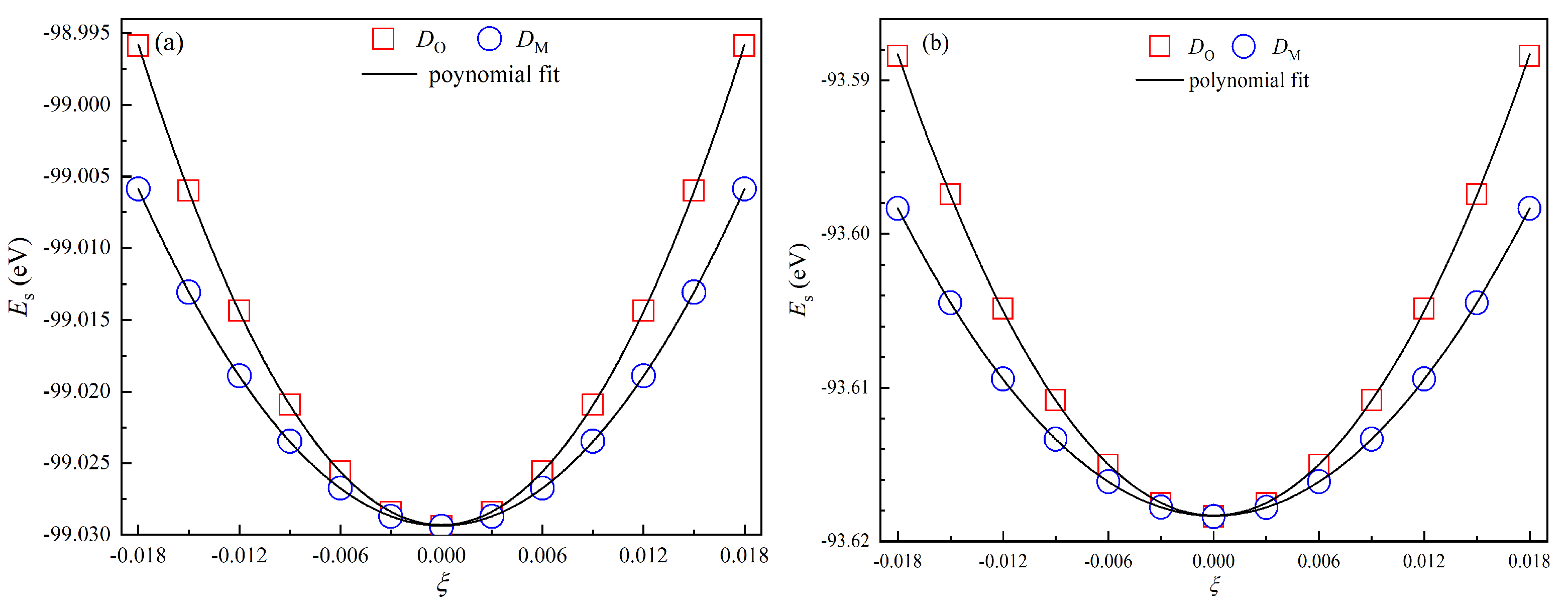
3.1. Structural and Elastic Properties
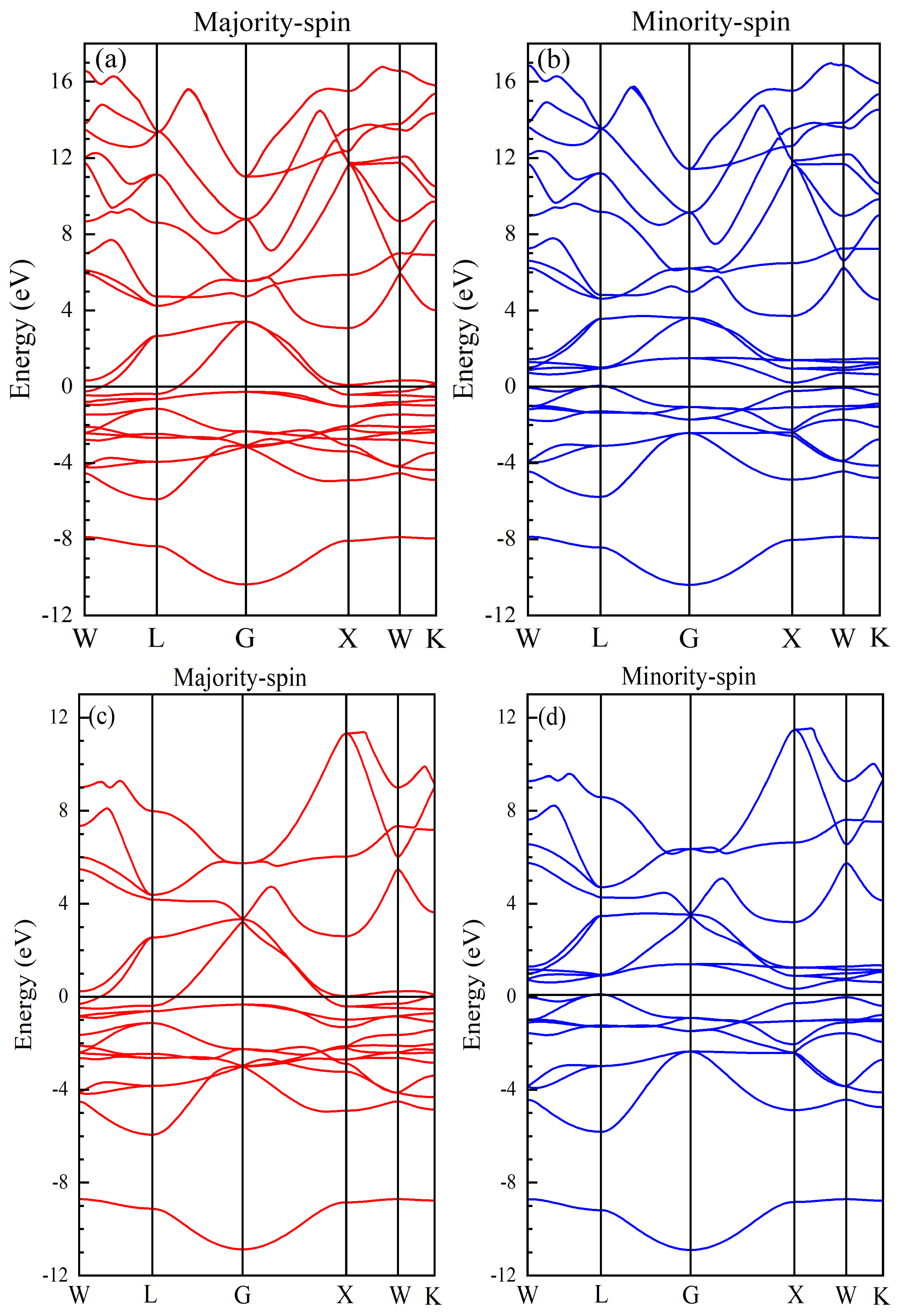
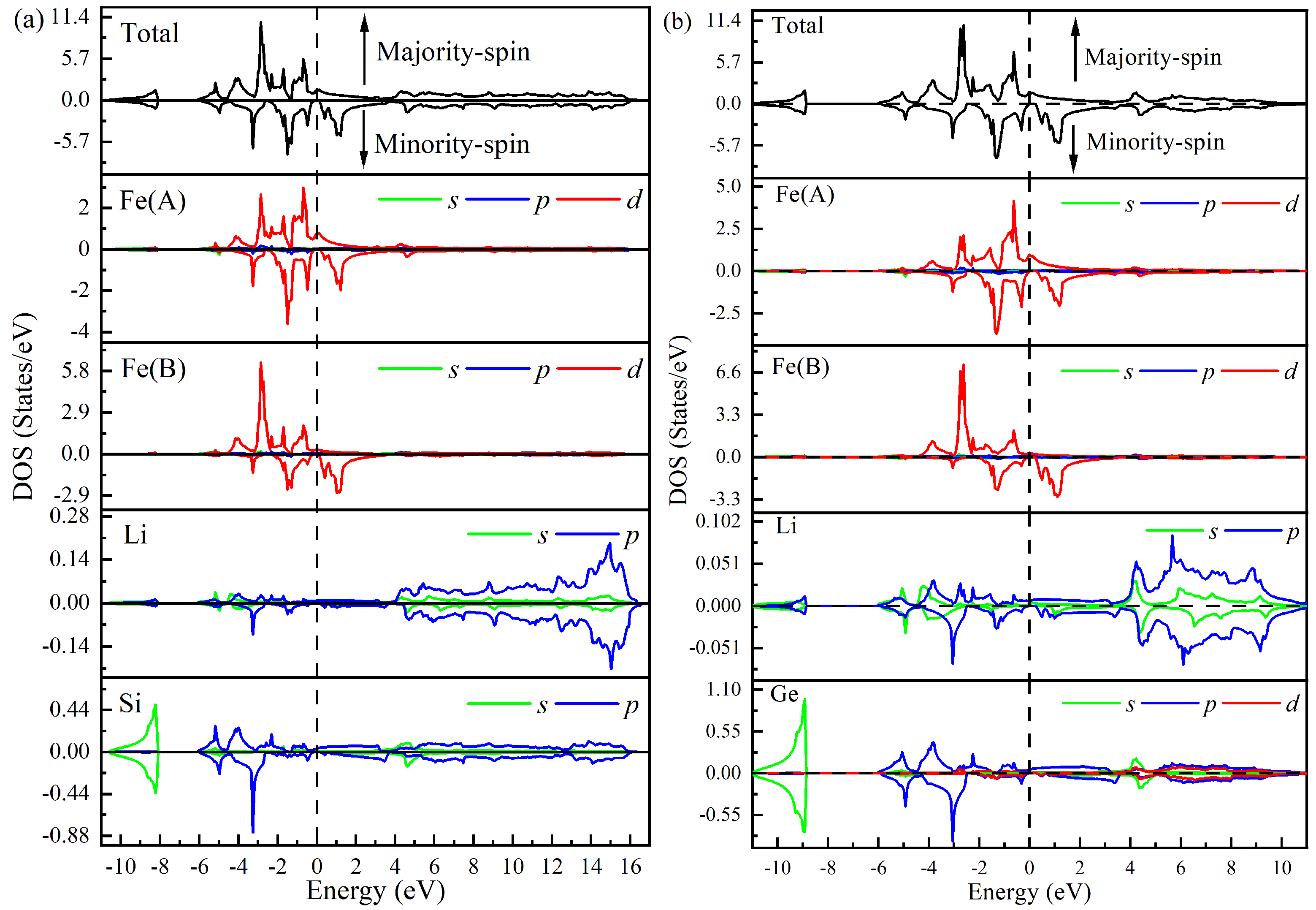
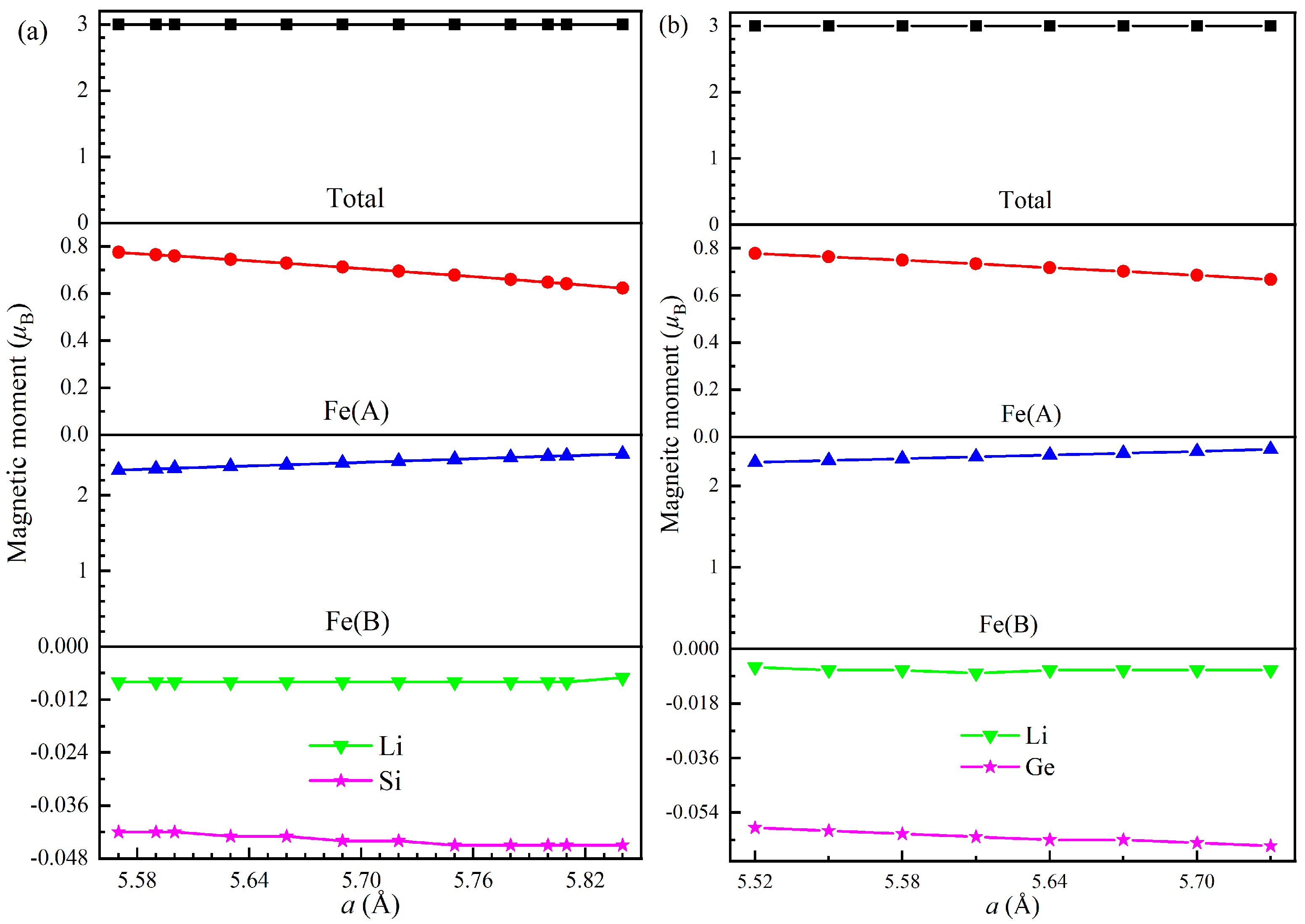
Electronic, Magnetic, and Half-Metallic Properties
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Tavares, S.; Yang, K.; Meyers, M.A. Heusler alloys: Past, properties, new alloys, and prospects. Prog. Mater. Sci. 2023, 132, 101017. [Google Scholar] [CrossRef]
- de Groot, R.A.; Mueller, F.M.; van Engen, P.G.; Buschow, K.H.J. New class of materials: Half-metallic ferromagnets. Phys. Rev. Lett. 1983, 50, 2024. [Google Scholar] [CrossRef]
- Li, G.; Li, Z.; Liu, Z. Study on the phase stability, mechanical and half-metallic properties of half-Heusler alloys FeMnZ (Z = Si, Ge, and Sn). J. Magn. Magn. Mater. 2025, 614, 172742. [Google Scholar] [CrossRef]
- Rachida, T.; Yahia, B.; Hichem, B.; Tayeb, B.E.; Said, M.; Ayoub, B.; Youcef, R.; Mohammed, R. Half-metallic ferromagnetism and thermoelectric performance of PdFeCrZ (Z = Al, Si, Sb, Ge) quaternary Heusler alloys for thermos-spintronic applications. J. Magn. Magn. Mater. 2025, 624, 173072. [Google Scholar] [CrossRef]
- Faid, F.; Mebarki, H.; Mokadem, K.; Abdalilah, F.M.; Benmakhlouf, A.; Khatiri, M.; Helaimia, T. Systematic study of structural, elastic, electronic, magnetism and half-metallic properties for the quaternary alloys: Heusler type VZrReZ (Z = Si, Ge and Sn). J. Magn. Magn. Mater. 2024, 605, 172345. [Google Scholar] [CrossRef]
- Mahmood, Q. Study of half metallic ferromagnetism, Curie temperature, and thermoelectric aspects of double perovskite oxides Ba2XMoO6 (X = Cr, Mn, Fe, Co) for spintronic applications. Mater. Sci. Semicon. Proc. 2025, 193, 109519. [Google Scholar] [CrossRef]
- Zhang, J.; Temnikov, F.; Ye, X.; Wang, X.; Pan, Z.; Liu, Z.; Pi, Z.; Tang, S.; Chen, C.; Pao, C. Large manipulation of ferrimagnetic curie temperature by A-site chemical substitution in ACu3Fe2Re2O12 (A = Na, Ca, and La) half metals. Inorg. Chem. 2025, 64, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Fu, J.; Xiang, Y.; Li, L.; Wu, X.; Wu, S. V doped orth-Ga2O3: Half-metallic ferromagnetism, large magnetic anisotropy energy and high curie temperature. J. Alloys Compd. 2025, 1010, 177301. [Google Scholar] [CrossRef]
- Mogulkoc, A.; Modarresi, M.; Rudenko, A.N. Two-dimensional chromium pnictides CrX (X = P, As, Sb): Half-metallic ferromagnets with high curie temperature. Phys. Rev. B 2020, 102, 024441. [Google Scholar] [CrossRef]
- Charifi, Z.; Guendouz, D.; Baaziz, H.; Soyalp, F.; Hamad, B. Ab-initio investigations of the structural, electronic, magnetic and mechanical properties of CrX (X = As, Sb, Se, and Te) transition metal pnictides and chalcogenides. Phys. Scr. 2019, 94, 14. [Google Scholar] [CrossRef]
- Kattan, N.A.; Ayyaz, A.; Alkhaldi, H.D.; Alkhaldi, N.D.; Bouzgarrou, S.; Boukhris, I.; Albalawi, H.; Mahmood, Q.; Al-Buriahi, M.S. First principles investigations of electronic, ferromagnetic, and thermoelectric aspects of chalcogenides BeCe2X4 (X = S, Se, Te) for spintronics. J. Phys. Chem. Solids 2025, 206, 112843. [Google Scholar] [CrossRef]
- Sohail, S.; Irfan, M.; Ain, Q.; Ibrahim, F.A.; Hamdy, M.; Issa, S.A.M.; Zakaly, H.M.H. First principles computation of exchange mechanism, radiation shielding, and physical properties of FeCu2SnX4 (X = S, Se, Te): Transitions metal based chalcogenides for spintronic and energy storage system applications. Mater. Sci. Semicon. Proc. 2025, 190, 109303. [Google Scholar] [CrossRef]
- Jain, V.K.; Lakshmi, N.; Jain, R.; Chandra, A.R. Electronic structure, elastic, magnetic, and optical Properties of Fe2MnZ (Z = Si, Ge, and Sn) full Heusler alloys: First-principle calculations. J. Supercond. Nov. Magn. 2019, 32, 739–749. [Google Scholar] [CrossRef]
- Ganai, Z.S.; Yousuf, S.; Batoo, K.M.; Khan, M.; Gupta, D.C. Half-metallicity and onsite Hubbard interaction on d-electronic states: A case study of Fe2NiZ (Z = Al, Ga, Si, Ge) Heusler systems. Philos. Mag. 2019, 99, 1551–1562. [Google Scholar] [CrossRef]
- Rai, D.P.; Rinkima, L.; Zuala, L.; Fomin, L.A.; Malikov, I.V.; Sayede, A.; Ghimire, M.P.; Thapa, R.K.; Zadeng, L. Pressure dependent half-metallic ferromagnetism in inverse Heusler alloy Fe2CoAl: DFT+U calculation. RSC Adv. 2020, 10, 44633–44640. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-W.; Hsing, C.-R.; Chang, C.-M.; Wei, C.-M. Electronic structures of 24-valence-electron full Heusler compounds investigated by density functional and GW calculations. J. Phys. Condens. Mat. 2020, 32, 175501. [Google Scholar] [CrossRef] [PubMed]
- Mansour, B.; Zoubir, A.; Ahmad, K.S.; Sabria, T.; Aftab, A.M.; Bouabdellah, B.; Abderrahim, B.M.; Amel, L. Electronic structure, thermoelectric, mechanical and phonon properties of full-Heusler alloy (Fe2CrSb): A first-principles study. Bull. Mater. Sci. 2021, 44, 221. [Google Scholar]
- Wang, L.; Cao, Y.; Zhang, C.; Xu, Y.; Zhou, S. Theoretical study of structural, mechanical, electronic, magnetic and thermodynamic properties of Cu2MnAl-type Fe2YAl (Y = Cr, Mo and W) full-Heusler alloys. Mater. Sci. Eng. B 2022, 278, 115639. [Google Scholar] [CrossRef]
- Erager, K.R.; Baigutlin, D.; Sokolovskiy, V.; Buchelnikov, V. On the possible half-metallic properties of Fe2RhZ (Z = Al, Si, Ga, Ge, In, Sn) ferromagnetic Husler alloys. J. Commun. Technol. Electron. 2023, 68, 389–399. [Google Scholar] [CrossRef]
- Bhattacharya, J.; Dutt, R.; Chakrabarti, A. Ab-initio predictions of mechanical, electronic, magnetic, and transport properties of bulk and heterostructure of a novel Fe-Cr based full Heusler chalcogenide. J. Phys. Chem. Solids 2023, 178, 111307. [Google Scholar] [CrossRef]
- Biswas, S.; Alagarsamy, P.; Srinivasan, A. Influence of atomic substitution on the structural stability and half-metallicity of Fe2-xCrxCoSi (x = 0 to 1) alloys. J. Magn. Magn. Mater. 2024, 612, 172648. [Google Scholar] [CrossRef]
- Hariharan, M.; Eithiraj, R.D. Structural, electronic, magnetic, and thermoelectric properties of newly predicted Fe2CoS and Ni2CoS alloys for spintronics applications: A DFT study. J. Magn. Magn. Mater. 2024, 589, 171553. [Google Scholar] [CrossRef]
- Pérez, C.A.S.; de Marchi, A.F. DFT calculations for electronic and magnetic properties of full Heusler Fe2MnAs alloy in perfect and defect structures. J. Magn. Magn. Mater. 2024, 589, 171541. [Google Scholar] [CrossRef]
- Iram, N.; Sharma, R.; Ahmed, J.; Almeer, R.; Kumar, A.; Abbas, Z. Exploring the physical, magnetic, opto-spintronics and thermoelectric properties of Fe2ZrAs Heusler alloy through DFT study. J. Phys. Chem. Solids 2025, 196, 112368. [Google Scholar] [CrossRef]
- Zaoui, Y.; Beldi, L.; Bouhafs, B.; Kanoun, M.B.; Goumri-Said, S. Dynamic stability, half-metallicity, and optical properties of Fe2CrX (X = Si, Ge) full-heusler alloys: Competition between L21 and XA ordering. Mater. Sci. Eng. B 2025, 317, 118168. [Google Scholar] [CrossRef]
- Jiang, D.; Ye, Y.; Gou, Q.; Wu, D.; Wen, Y. First-principles predictions on structural, elastic and half-metallic properties Fe2LiAs Heusler compound. J. Mag. Mag. Mater. 2018, 458, 235–240. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kresse, G.; Fürthmller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Fürthmller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Francis, B. Finite elastic strain of cubic crystals. Phys. Rev. 1947, 71, 809–824. [Google Scholar]
- Kirklin, S.; Thompson, A.; Doak, J.W.; Aykol, M.; Rühl, S.; Wolverton, C. The Open Quantum Materials Database (OQMD): Assessing the accuracy of DFT formation energies. Npj Comput. Mater. 2015, 1, 15010. [Google Scholar] [CrossRef]
- Wu, S.-C.; Fecher, C.H.; Naghavi, S.S.; Felser, C. Elastic properties and stability of Heusler compounds: Cubic Co2YZ compounds with L21 structure. J. Appl. Phys. 2019, 125, 082523. [Google Scholar] [CrossRef]
- Hill, R. The elastic behaviour of a crystalline aggregate. Proc. Phys. Soc. A 1952, 65, 349–354. [Google Scholar] [CrossRef]
- Voigt, W. Lehrbuch der Kristallphysik; Taubner: Leipzig, Germany, 1928. [Google Scholar]
- Reuss, A. Calculation of the flow limits of mixed crystals on the basis of the plasticity of monocrystals. Z. Angew. Math. Mech. 1929, 9, 49–58. [Google Scholar] [CrossRef]
- Frantsevich, I.N.; Voronov, F.F.; Bokuta, S.A. Elastic Constants and Elastic Moduli of Metals and Insulators Handbook; Naukova Dumka: Kiev, Ukraine, 1983; pp. 60–180. [Google Scholar]
- Anderson, O.L. A simplified method for calculating the debye temperature from elastic constants. J. Phys. Chem. Solids 1963, 24, 909–917. [Google Scholar] [CrossRef]
- Schreiber, E.; Anderson, O.L.; Soga, N. Elastic Constants and Their Measurements; McGraw: New York, NY, USA, 1973. [Google Scholar]
- Belomestnykh, V.N. The acoustical Grüneisen constants of solids. Tech. Phys. Lett. 2004, 30, 91–93. [Google Scholar] [CrossRef]
- Fine, M.E.; Brown, L.D.; Marcus, H.L. Elastic constants versus melting temperature in metals. Scr. Metall. 1984, 18, 951. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Z | B | Ref. | |||||
|---|---|---|---|---|---|---|---|
| Si | 5.590 | 156.24 | 4.157 | 219.48 | 124.62 | 132.96 | This |
| 5.561 | [34] | ||||||
| Ge | 5.702 | 141.35 | 4.679 | 194.71 | 114.67 | 106.73 | This |
| 5.684 | [34] |
| Z | B | G | E | |
|---|---|---|---|---|
| Si | 156.24 | 87.99 | 222.26 | 0.2629 |
| Ge | 141.35 | 80.05 | 184.74 | 0.2822 |
| Z | |||||||
|---|---|---|---|---|---|---|---|
| Si | 5.588 | 6997 | 3968 | 4412 | 559.3 | 1.432 | 1850 |
| Ge | 6.859 | 5883 | 3241 | 3612 | 449.0 | 1.528 | 1704 |
| Z | Total | Fe(A) | Fe(B) | Li | Z |
|---|---|---|---|---|---|
| Si | 3.000 | 0.765 | 2.351 | −0.008 | −0.042 |
| Ge | 3.000 | 0.684 | 2.424 | −0.007 | −0.064 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, Y.; Yu, Y.; Lai, Z.; Zeng, X. Structural, Elastic, Electronic, Magnetic, and Half-Metallic Properties of Full-Heusler Compounds Fe2LiZ (Z = Ge and Si): A First-Principles Study. Metals 2025, 15, 808. https://doi.org/10.3390/met15070808
Wen Y, Yu Y, Lai Z, Zeng X. Structural, Elastic, Electronic, Magnetic, and Half-Metallic Properties of Full-Heusler Compounds Fe2LiZ (Z = Ge and Si): A First-Principles Study. Metals. 2025; 15(7):808. https://doi.org/10.3390/met15070808
Chicago/Turabian StyleWen, Yufeng, Yanlin Yu, Zhangli Lai, and Xianshi Zeng. 2025. "Structural, Elastic, Electronic, Magnetic, and Half-Metallic Properties of Full-Heusler Compounds Fe2LiZ (Z = Ge and Si): A First-Principles Study" Metals 15, no. 7: 808. https://doi.org/10.3390/met15070808
APA StyleWen, Y., Yu, Y., Lai, Z., & Zeng, X. (2025). Structural, Elastic, Electronic, Magnetic, and Half-Metallic Properties of Full-Heusler Compounds Fe2LiZ (Z = Ge and Si): A First-Principles Study. Metals, 15(7), 808. https://doi.org/10.3390/met15070808