

Towards Using MMO Anodes in Zinc Electrorefining: Mn Removal by Simulated Plant Off-Gas

 ,
,
Abstract
:1. Introduction
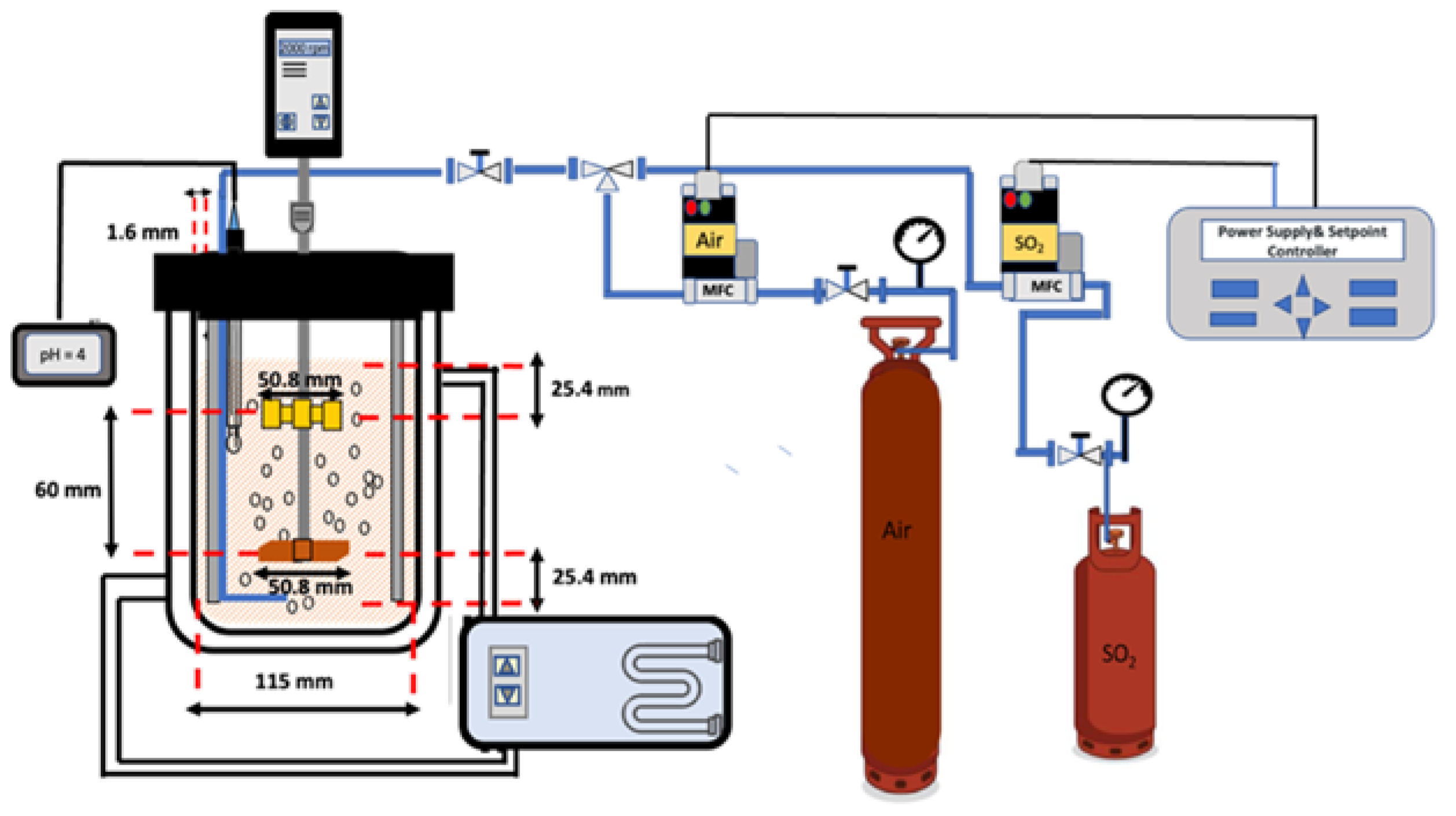
2. Materials and Methods
2.1. Materials and Reagents
2.2. Methods
2.3. Analysis and Characterization
3. Theory (Reaction Mechanisms)
3.1. Initiation
3.2. Manganese-Sulfite Complex Formation
3.3. Peroxymonosulfate Radical Generation
3.4. Regeneration of Mn3+ as a Catalyst
3.5. Manganese Dioxide Precipitation
4. Results
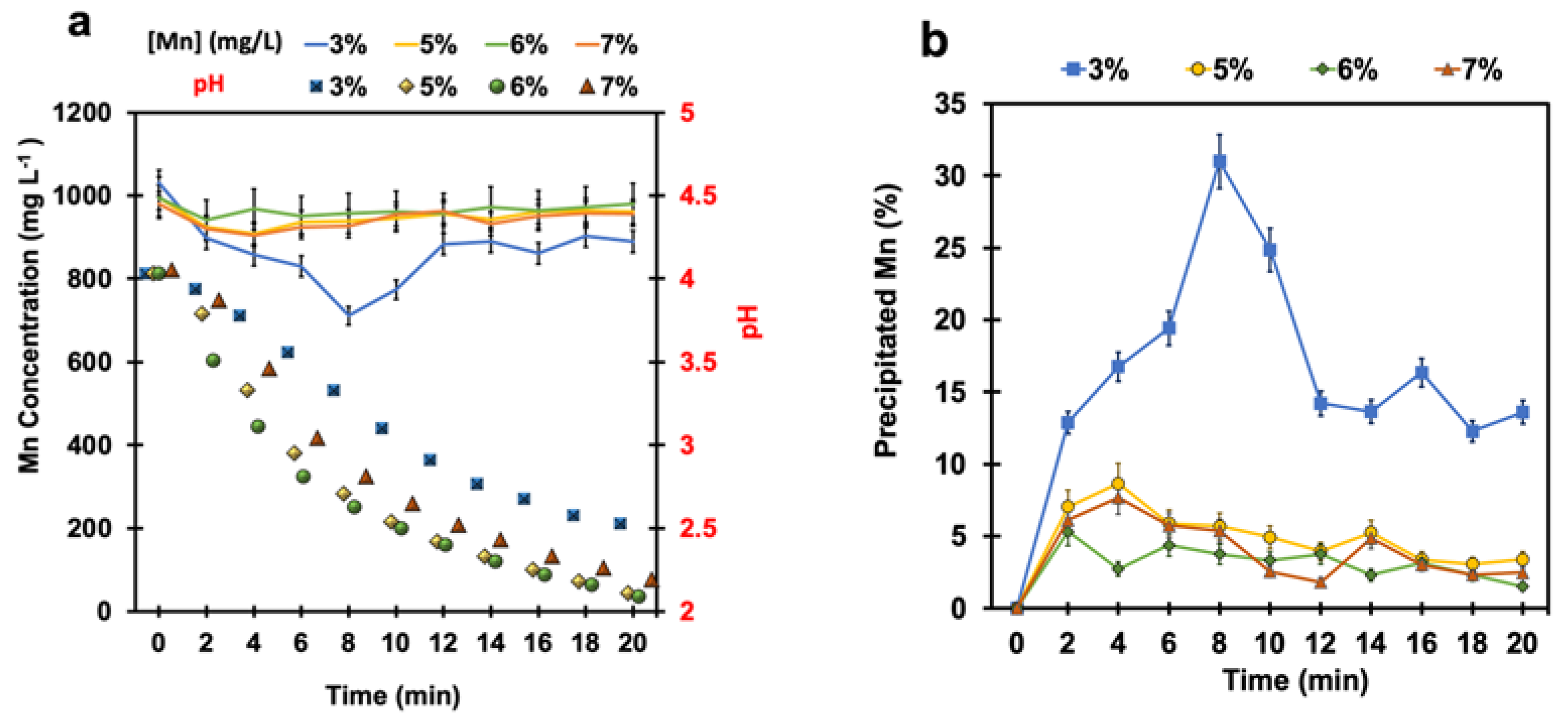
4.1. Manganese Removal without pH Adjustment
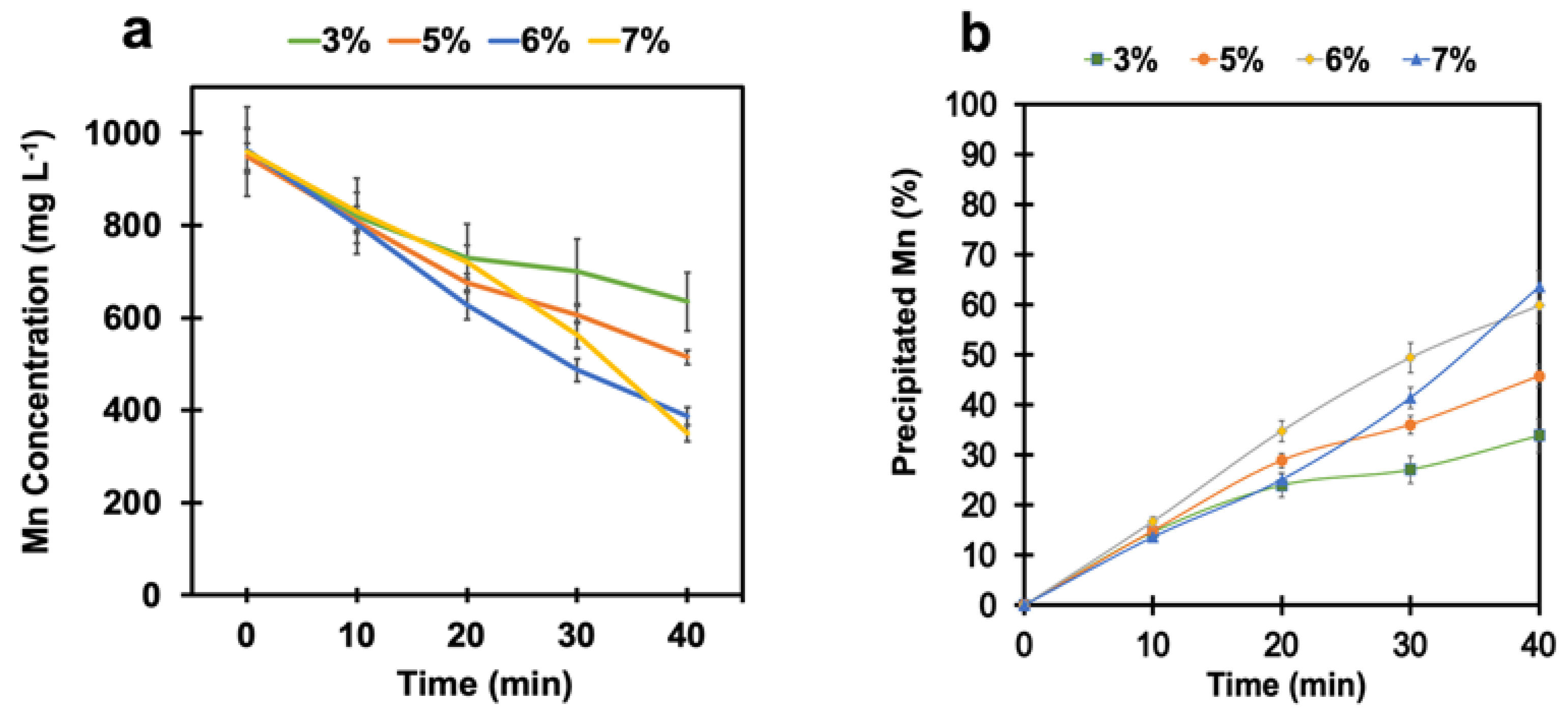
4.2. Manganese Removal with pH Adjustment
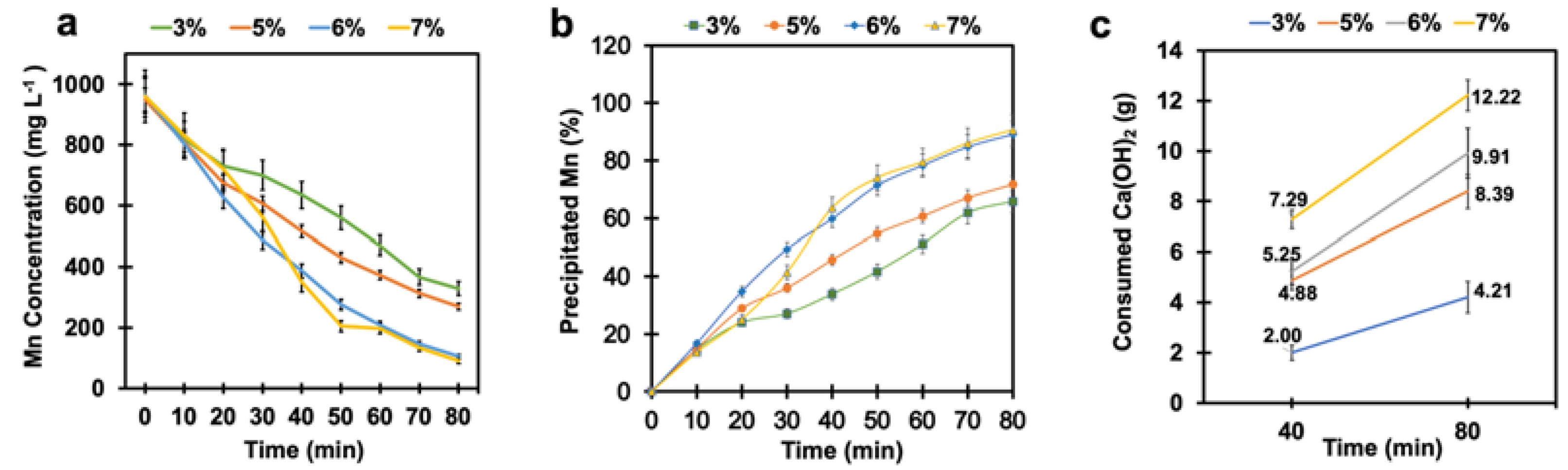
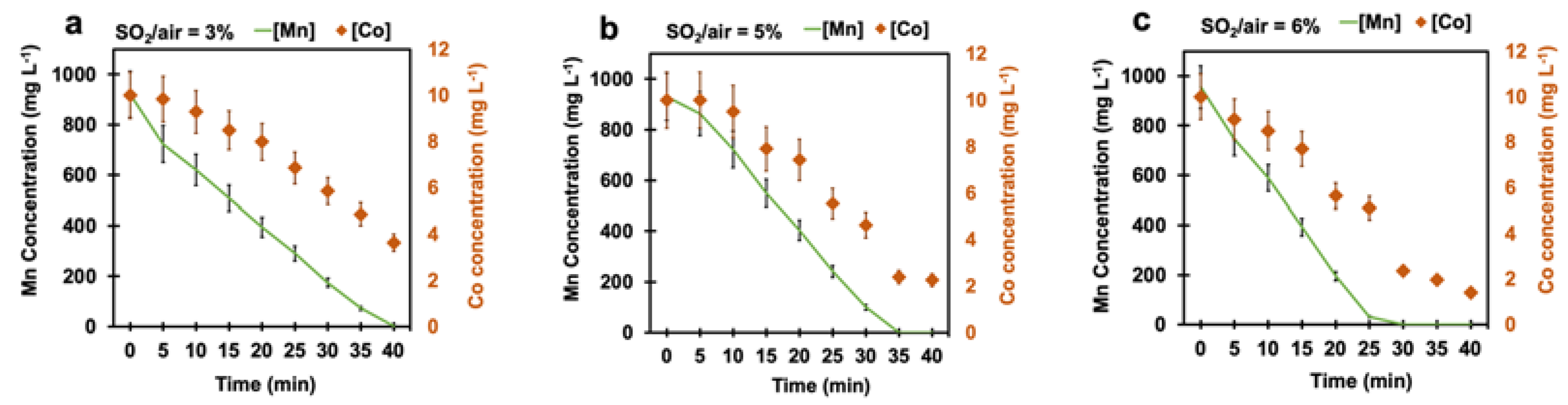
4.3. Effect of Time
4.4. Effect of Cobalt on Manganese Removal
4.4.1. Cobalt as Catalyst
4.4.2. Cobalt Removal
4.4.3. Effect of Cobalt Concentration on Manganese Removal
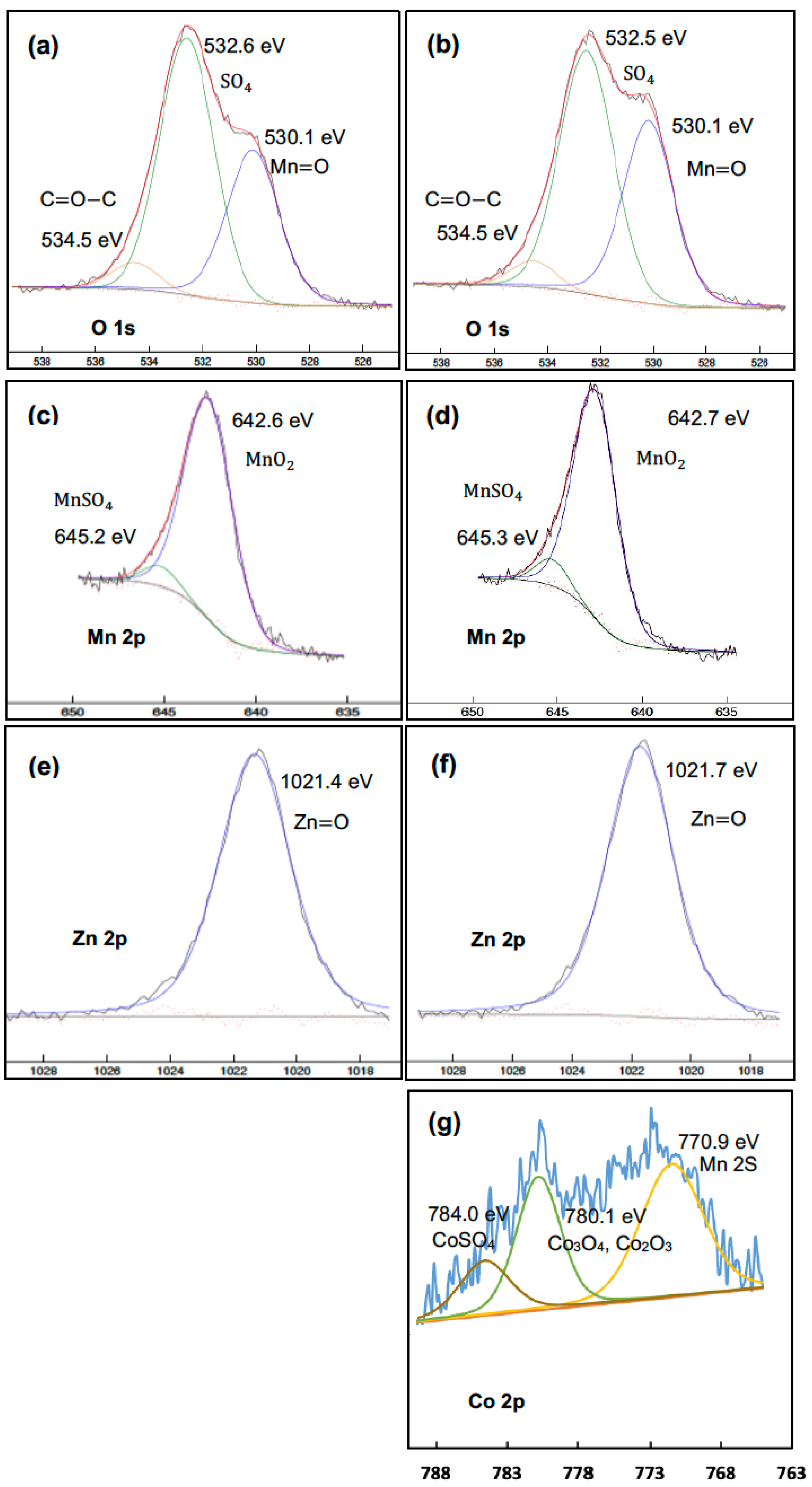
4.5. Characterization of the Precipitates
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- He, S.; Xu, R.; Sun, L.; Fan, Y.; Zhao, Z.; Liu, H.; Lv, H. Electrochemical characteristics of Co3O4-doped β-PbO2 composite anodes used in long-period zinc electrowinning. Hydrometallurgy 2020, 194, 105357. [Google Scholar] [CrossRef]
- Jaimes, R.; Miranda-Hernández, M.; Lartundo-Rojas, L.; González, I. Characterization of anodic deposits formed on Pb–Ag electrodes during electrolysis in mimic zinc electrowinning solutions with different concentrations of Mn(II). Hydrometallurgy 2015, 156, 53–62. [Google Scholar] [CrossRef]
- Sinclair, R.J. The Extractive Metallurgy of Zinc; AusIMM: Carlton, VIC, Australia, 2005. [Google Scholar]
- Wu, F.; Yang, Z.-Q.; Sun, W.; Chen, X.; Qi, H.; Wang, L.-D. Electrochemical Properties of Ti/SnO2-Sb-Ir Electrodes Doped with a Low Iridium Content for the Oxygen Evolution Reaction in an Acidic Environment. Ind. Eng. Chem. Res. 2022. [Google Scholar] [CrossRef]
- Ye, W.; Xu, F.; Jiang, L.; Duan, N.; Li, J.; Ma, Z.; Zhang, F.; Chen, L. Lead release kinetics and film transformation of Pb-MnO2 pre-coated anode in long-term zinc electrowinning. J. Hazard. Mater. 2021, 408, 124931. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; He, Y.; Li, W.; Wang, Z.; Yu, L.; Jiang, Y.; Liu, X.; Kang, J.; Gao, H.; Lin, N. Insight into electrochemical performance of porous FexSiy intermetallic anode for zinc electrowinning. Mater. Des. 2020, 191, 108645. [Google Scholar] [CrossRef]
- Trasatti, S. Electrocatalysis: Understanding the success of DSA®. Electrochim. Acta 2000, 45, 2377–2385. [Google Scholar] [CrossRef]
- Krstić, V.; Pešovski, B. Reviews the research on some dimensionally stable anodes (DSA) based on titanium. Hydrometallurgy 2019, 185, 71–75. [Google Scholar] [CrossRef]
- Ma, D.; Ngo, V.; Raghavan, S.; Sandoval, S. Degradation of Ir-Ta oxide coated Ti anodes in sulfuric acid solutions containing fluoride. Corros. Sci. 2020, 164, 108358. [Google Scholar] [CrossRef]
- Zhang, W.; Cheng, C.Y. Manganese metallurgy review. Part III: Manganese control in zinc and copper electrolytes. Hydrometallurgy 2007, 89, 178–188. [Google Scholar] [CrossRef]
- Mahon, M.; Alfantazi, A. Manganese consumption during zinc electrowinning using a dynamic process simulation. Hydrometallurgy 2014, 150, 184–191. [Google Scholar] [CrossRef]
- Zhang, W.; Ghali, E.; Houlachi, G. Review of oxide coated catalytic titanium anodes performance for metal electrowinning. Hydrometallurgy 2017, 169, 456–467. [Google Scholar] [CrossRef]
- de Menezes, D.R. Performance Evaluation of Mixed Metal Oxide Anodes for Zinc Electrowinning. Ph.D. Thesis, Université Laval, Québec, QC, Canada, 2021. [Google Scholar]
- Ménard, V. Controlled Oxidative Precipitation of Manganese from an Industrial Zinc Sulphate Solution Using a Sulphur Dioxide and Oxygen Gas Mixture. Master’s Thesis, McGill University, Montréal, QC, Canada, 2004. [Google Scholar]
- Patil, D.S.; Chavan, S.M.; Oubagaranadin, J.U.K. A review of technologies for manganese removal from wastewaters. J. Environ. Chem. Eng. 2016, 4, 468–487. [Google Scholar] [CrossRef]
- Rethinaraj, J.P.; Visvanathan, S. Preparation and properties of electrolyc manganese dioxide. J. Power Sources 1993, 42, 335–343. [Google Scholar] [CrossRef]
- Awual, M.R. A novel facial composite adsorbent for enhanced copper (II) detection and removal from wastewater. Chem. Eng. J. 2015, 266, 368–375. [Google Scholar] [CrossRef]
- Awual, M.R.; Hasan, M.M.; Rahman, M.M.; Asiri, A.M. Novel composite material for selective copper (II) detection and removal from aqueous media. J. Mol. Liq. 2019, 283, 772–780. [Google Scholar] [CrossRef]
- Orue, B.P.; Botelho Junior, A.B.; Tenório, J.A.S.; Espinosa, D.C.R.; Baltazar, M.d.P.G. Kinetic study of manganese precipitation of nickel laterite leach based-solution by ozone oxidation. Ozone Sci. Eng. 2021, 43, 324–338. [Google Scholar] [CrossRef]
- Mulaudzi, N.; Mahlangu, T. Oxidative precipitation of Mn (II) from cobalt leach solutions using dilute SO2/air gas mixture. J. South. Afr. Inst. Min. Metall. 2009, 109, 375–382. [Google Scholar]
- Bello-Teodoro, S.; Pérez-Garibay, R.; Uribe-Salas, A. The controlled oxidative precipitation of manganese oxides from Mn(II) leach liquors using SO2/air gas mixtures. Miner. Eng. 2011, 24, 1658–1663. [Google Scholar] [CrossRef]
- Teixeira, L.A.C.; Queiroz, J.P.L.; Marquez-Sarmiento, C. Oxidative precipitation of manganese from dilute waters. Mine Water Environ. 2017, 36, 452–456. [Google Scholar] [CrossRef]
- Saidi, M.; Kadkhodayan, H. Toxic heavy metal removal from sulfide ores using potassium permanganate: Process development and waste management. J. Environ. Manag. 2020, 276, 111354. [Google Scholar] [CrossRef]
- Zhang, W.; Singh, P.; Muir, D. Oxidative precipitation of manganese with SO2/O2 and separation from cobalt and nickel. Hydrometallurgy 2002, 63, 127–135. [Google Scholar] [CrossRef]
- Menard, V.; Demopoulos, G.P. Gas transfer kinetics and redox potential considerations in oxidative precipitation of manganese from an industrial zinc sulphate solution with SO2/O2. Hydrometallurgy 2007, 89, 357–368. [Google Scholar] [CrossRef]
- Brandt, C.; Van Eldik, R. Transition metal-catalyzed oxidation of sulfur (IV) oxides. Atmospheric-relevant processes and mechanisms. Chem. Rev. 1995, 95, 119–190. [Google Scholar] [CrossRef]
- Pérez-Garibay, R.; Bello-Teodoro, S.; Rojas-Montes, J.C. Thermodynamic simulation of the reaction mechanism of Mn2+ oxidation with an SO2/O2 mixture. Hydrometallurgy 2018, 176, 260–265. [Google Scholar] [CrossRef]
- Zhang, W.; Singh, P.; Muir, D. SO2/O2 as an oxidant in hydrometallurgy. Miner. Eng. 2000, 13, 1319–1328. [Google Scholar] [CrossRef]
- Coichev, N.; Van Eldik, R. Kinetics and mechanism of the sulfite-induced autoxidation of cobalt(II) in aqueous azide medium. Inorg. Chem. 1991, 30, 2375–2380. [Google Scholar] [CrossRef]
- Ghanbari, F.; Moradi, M. Application of peroxymonosulfate and its activation methods for degradation of environmental organic pollutants. Chem. Eng. J. 2017, 310, 41–62. [Google Scholar] [CrossRef]
- Xiao, S.; Cheng, M.; Zhong, H.; Liu, Z.; Liu, Y.; Yang, X.; Liang, Q. Iron-mediated activation of persulfate and peroxymonosulfate in both homogeneous and heterogeneous ways: A review. Chem. Eng. J. 2020, 384, 123265. [Google Scholar] [CrossRef]
- Li, H.; Zhao, Z.; Qian, J.; Pan, B. Are Free Radicals the Primary Reactive Species in Co(II)-Mediated Activation of Peroxymonosulfate? New Evidence for the Role of the Co(II)–Peroxymonosulfate Complex. Environ. Sci. Technol. 2021, 55, 6397–6406. [Google Scholar] [CrossRef]
- Hou, J.; He, X.; Zhang, S.; Yu, J.; Feng, M.; Li, X. Recent advances in cobalt-activated sulfate radical-based advanced oxidation processes for water remediation: A review. Sci. Total Environ. 2021, 770, 145311. [Google Scholar] [CrossRef]
- Anipsitakis, G.P.; Dionysiou, D.D. Radical generation by the interaction of transition metals with common oxidants. Environ. Sci. Technol. 2004, 38, 3705–3712. [Google Scholar] [CrossRef] [PubMed]
- Brandt, C.; Van Eldik, R. The formation of dithionate during the iron(III)-catalysed autoxidation of sulfur(IV)-oxides. Atmos. Environ. 1997, 31, 4247–4249. [Google Scholar] [CrossRef]
- Anipsitakis, G.P.; Dionysiou, D.D. Degradation of organic contaminants in water with sulfate radicals generated by the conjunction of peroxymonosulfate with cobalt. Environ. Sci. Technol. 2003, 37, 4790–4797. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Li, Q.; Wang, Q.; Yan, X.; Li, B.; Feng, L.; Wu, C.; Qiu, R.; Zhang, H.; Yang, Z.; et al. A review on the transformation of birnessite in the environment: Implication for the stabilization of heavy metals. J. Environ. Sci. 2023, 139, 496–515. [Google Scholar] [CrossRef]
- Hinkle, M.A.G.; Dye, K.G.; Catalano, J.G. Impact of Mn(II)-Manganese Oxide Reactions on Ni and Zn Speciation. Environ. Sci. Technol. 2017, 51, 3187–3196. [Google Scholar] [CrossRef] [PubMed]
- Peña, J.; Bargar, J.R.; Sposito, G. Copper sorption by the edge surfaces of synthetic birnessite nanoparticles. Chem. Geol. 2015, 396, 196–207. [Google Scholar] [CrossRef]
- Xu, T.; Roepke, E.W.; Flynn, E.D.; Rosenfeld, C.E.; Balgooyen, S.; Ginder-Vogel, M.; Schuler, C.J.; Santelli, C.M. Aqueous Co removal by mycogenic Mn oxides from simulated mining wastewaters. Chemosphere 2023, 327, 138467. [Google Scholar] [CrossRef]
- Moulder, J.F.; Chastain, J. Handbook of X-ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data; Physical Electronics Division, Perkin-Elmer Corporation: Waltham, MA, USA, 1992. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | Green Frame | Brown Frame | Yellow Frame | Orange Frame | Blue Frame | Purple Frame |
|---|---|---|---|---|---|---|
| Mn | 62.7 | 28.3 | 21.8 | 0.7 | 25.4 | 4.1 |
| Zn | 7.8 | 25.7 | 22.9 | 3.2 | 27.7 | 9.1 |
| O | 26.6 | 32.8 | 42.7 | 62.8 | 39.1 | 51.6 |
| Ca | – | – | 3.4 | 17.7 | 0.7 | 16.3 |
| Co | – | – | – | – | 0.1 | – |
| S | 2.9 | 13.2 | 9.2 | 15.6 | 7 | 18.9 |
| Element | Without Co Addition (%) | With 10 mg L−1 Co Addition (%) |
|---|---|---|
| O1s | 54.7 | 53.1 |
| C1s | 17.1 | 19.7 |
| Ca 2p | 8.2 | 6.3 |
| Zn 2p | 7.4 | 8.4 |
| S 2p | 6.4 | 5.6 |
| Co 2p | – | 0.1 |
| Mn 2p | 6.2 | 6.8 |
| Element | keV | Line | Concentration of Precipitates without Cobalt Addition (%) | Concentration of Precipitates with 10 mg L−1 Co Addition (%) |
|---|---|---|---|---|
| Mn | 5.8 | 6.2 | 6.79 | |
| Zn | 8.6 | 7.1 | 9 | |
| Co | 6.9 | 0 | 0.3 | |
| Ca | 3.6 | 26.14 | 20.87 | |
| S | 2.3 | 16.66 | 14.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Askarian, M.; Mousavi, F.; Mollaabbasi, R.; Benguerel, E.; Brown, C.; Houlachi, G.; Alamdari, H. Towards Using MMO Anodes in Zinc Electrorefining: Mn Removal by Simulated Plant Off-Gas. Metals 2023, 13, 1675. https://doi.org/10.3390/met13101675
Askarian M, Mousavi F, Mollaabbasi R, Benguerel E, Brown C, Houlachi G, Alamdari H. Towards Using MMO Anodes in Zinc Electrorefining: Mn Removal by Simulated Plant Off-Gas. Metals. 2023; 13(10):1675. https://doi.org/10.3390/met13101675
Chicago/Turabian StyleAskarian, Masoomeh, Fariba Mousavi, Roozbeh Mollaabbasi, Elyse Benguerel, Carl Brown, Georges Houlachi, and Houshang Alamdari. 2023. "Towards Using MMO Anodes in Zinc Electrorefining: Mn Removal by Simulated Plant Off-Gas" Metals 13, no. 10: 1675. https://doi.org/10.3390/met13101675
APA StyleAskarian, M., Mousavi, F., Mollaabbasi, R., Benguerel, E., Brown, C., Houlachi, G., & Alamdari, H. (2023). Towards Using MMO Anodes in Zinc Electrorefining: Mn Removal by Simulated Plant Off-Gas. Metals, 13(10), 1675. https://doi.org/10.3390/met13101675